University of Illinois, Dept of Biochemistry
How Does Estrogen Induce Proliferation of Cancer Cells?
SMALL MOLECULE INHIBITORS OF ESTROGEN AND PROGESTERONE RECEPTOR ACTION IN BREAST CANCER CELLS
SMALL MOLECULE INHIBITORS OF ANDROGEN RECEPTOR ACTION IN PROSTATE CANCER CELLS
REGULATION OF IMMUNE SURVEILLANCE AND APOPTOSIS BY PROTEINASE INHIBITOR 9
HOW DOES ESTROGEN INDUCE PROLIFERATION OF CANCER CELLS?
Reseachers: Mathew Cherian and Neal Andruska
Within the steroid/nuclear family of receptors, estrogen receptor (ER) and androgen receptor (AR) are unusual in their ability to stimulate cell proliferation. The ability of activated ER and AR to induce cell proliferation is central to their roles in breast and prostate cancer. Although drugs that prevent ER and AR from working are mainstays in the treatment of breast and prostate cancer, and more than 50,000 scientific papers have been published on estrogen receptor, as the eminent cancer biologist Bob Weinberg stated in his classic book The Biology of Cancer, “breast cancer…. has been the subject of extensive research, yet we still have only a vague understanding at the molecular level of how the growth of these tumors is controlled.”
Why do we care?
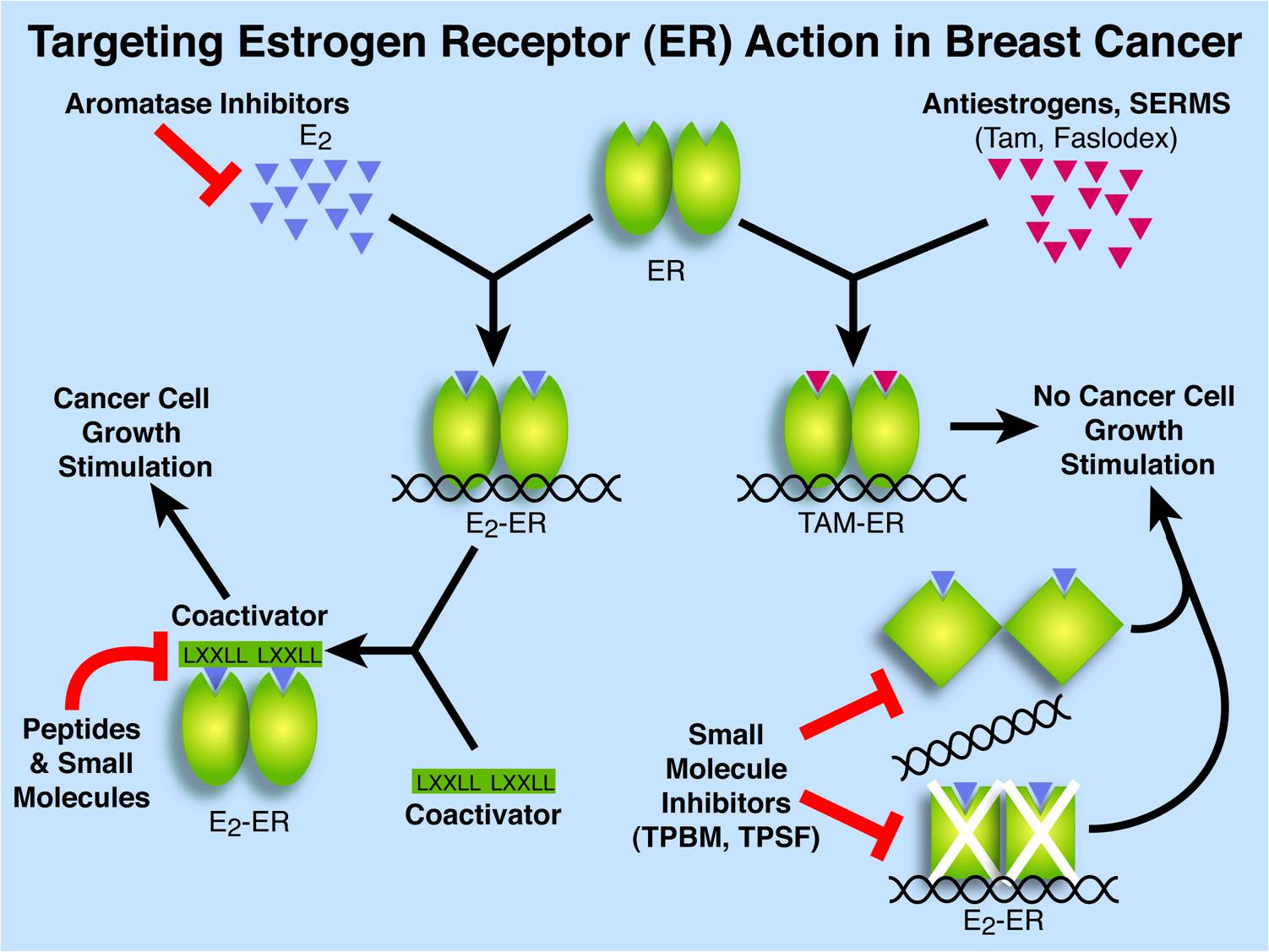
Breast cancer is the second leading cause of cancer deaths among American women. At the time of diagnosis about 2/3 of human breast cancers are ER positive. Estrogens stimulate the proliferation and metastatic potential of these early stage breast cancers. Successful treatment strategies for these ER-positive cancers involve blocking ER action; either by inhibiting estrogen production using aromatase inhibitors, or by interfering with binding of estrogens to ER using selective estrogen receptor modulators (SERMs) such as tamoxifen and Faslodex/Fulvestrant/ICI 182,780 that compete with estrogens for binding in the ER&alpha ligand binding pocket. The successful therapeutic use of SERMs and aromatase inhibitors demonstrates a central role for estrogens in the development and progression of breast cancer. Although, several candidate proteins and sites have been proposed, the precise mechanisms by which estrogens stimulate proliferation of breast cancer cells is largely unresolved.
While estrogen production and ER action are key therapeutic targets, breast cancers eventually develop resistance. Identifying key genes and pathways used by estrogens bound to ER to induce cell proliferation will likely lead to new prognostic indicators, provide a new generation of therapeutic targets and enhance the development of a new generation of ER inhibitors that target its ability to induce cell proliferation without interfering with potentially beneficial functions of ER.
To evaluate possible sites at which estrogen induces cell proliferation, we need to consider how estrogens and ER work.
How do estrogens work?
At the molecular level, estrogens, such as 11β-estradiol (E2), bind to the protein Estrogen Receptor α (ERα) and can act via several pathways. (A second form of ER, ERα, is often absent in ERα-positive breast cancer cells and has been proposed to exert anti-proliferative effects.) The most extensively characterized action of ERα is the direct regulation of gene expression in the nucleus. In response to high affinity estrogen binding, ER&alpha homodimerizes and translocates to the nucleus where it binds specific DNA sequences, termed estrogen response elements, or is tethered to DNA via interaction with other regulatory proteins bound to their DNA recognition sequences.
ERα undergoes a conformational change upon ligand binding that facilitates the recruitment of coactivators. Bound coactivators promote assembly of a multiprotein complex that enables chromatin remodeling and stabilization of an active transcription complex. When tamoxifen or other SERMs are bound to ER instead of E2, the ER often recruits corepressors instead of coactivators.
The E2-ERα complex can also act in the cytosol to rapidly activate several membrane-associated protein kinase-based signaling pathways, including the pro-proliferation ERK1/2 signaling pathway. However, ER activation of the ERK1/2 is not sufficient to induce cell proliferation, and at least some ER must enter the nucleus to trigger cell proliferation.
A third pathway of estrogen action that is independent of ER, based on binding of estrogens to membrane protein GPR30 has been implicated in estrogen mediated signaling, but remains controversial. Its reported responses to antiestrogens do not support a key role of GPR30 in cell proliferation and a mouse knockout of GPR30 had no obvious phenotypic alterations.
Although not experimentally proven, most researchers support the idea that estrogens, acting via binding to ERα, trigger cell proliferation by regulating expression of key cell cycle control genes. We believe that clear answers to this question have been difficult to achieve because most researchers attacked the problem using a single method or approach. To address the old question of how estrogens induce cell proliferation in new ways, we developed a toolkit containing a series of novel individual approaches that together create a new way to look at this important problem. Two of these new approaches can be described here.
How do estrogens induce cell proliferation? New tools for an old question.
We identified a clonal cell line, derived from a cancer cell line whose proliferation is strongly stimulated by estrogen. This clonally derived cell line has completely lost the capacity for estrogen-stimulated proliferation. Surprisingly, this cell line contains similar levels of ER as the parental cell line and, in limited studies, retains the ability to induce expression of ER target genes. Identifying changes in the pattern of gene expression in these cells, or alterations in other pathways compared to the nearly isogenic parental cells, will shed new light on how estrogen-stimulates cell proliferation.
In our efforts to develop small molecule inhibitors of ER action, we identified small molecule inhibitors of ER that block estrogen-stimulated cell proliferation, but have different abilities to inhibit expression of individual E2-regulated genes. Integrating these and other complementary tools provides a unique capability to identify key genes and pathways that trigger E2-stimulated proliferation of breast, ovarian and uterine cancer cells.
Selected Publications
Mao C, Livezey M, Kim JE and Shapiro DJ (2016) Antiestrogen Resistant Cell Lines Expressing Estrogen Receptor α Mutations Upregulate the Unfolded Protein Response and are Killed by BHPI. Scientific Reports, 6:34753. PMCID: 5054422.