University of Illinois, Dept of Biochemistry
HOW DOES ESTROGEN INDUCE PROLIFERATION OF CANCER CELLS?
Small Molecule Inhibitors of Estrogen and Progesterone Receptor Action in Breast Cancer Cells
SMALL MOLECULE INHIBITORS OF ANDROGEN RECEPTOR ACTION IN PROSTATE CANCER CELLS
REGULATION OF IMMUNE SURVEILLANCE AND APOPTOSIS BY PROTEINASE INHIBITOR 9
SMALL MOLECULE INHIBITORS OF ESTROGEN AND PROGESTERONE RECEPTOR ACTION IN BREAST CANCER CELLS
Collaborators: Professors Paul Hergenrother, William Helferich, Emad Tajkhorshid, Timothy Fan and Claudio Grosman (UIUC), Steve Nordeen (Univ Colorado Health Sciences Center), Elizabeth Wilson (Univ N. Carolina), Martha Stampfer (Lawrence Berkeley National Lab)
Alumni: Dr. Stanley Wang (MD/PhD), Dr. Chen Zhang, Dr. Irene Aninye, Dr. Milu Cherian, Nicole Patterson-Kretzer
Researchers: Neal Andruska, Mathew Cherian, Xiaobin Zheng, Liqun Yu, Dr. Chengjian Mao
Estrogens, acting via estrogen receptor (ER), stimulate cell proliferation, and tumor growth. Most ovarian, breast and endometrial cancers are ER positive. Although initially effective in ER positive breast cancer, endocrine therapy to block estrogen production and use of antiestrogens that compete with estrogens for binding to ERa often results in development of resistance. Most ovarian cancers present at an advanced stage. Although these tumors are usually ER positive, endocrine therapy targeting estrogen production, or estrogen binding to ER, is usually ineffective and the tumors are treated with chemotherapy. Although most tumors are initially responsive to combination chemotherapy, tumors recur as resistant ovarian cancer and approximately two thirds of patients die within 5 years. The presence of ERa in many therapy-resistant breast and ovarian cancers suggested the presence of additional modes of ERa action potentially targetable with small molecule biomodulators. We developed and implemented screens to identify novel small molecule inhibitors targeting previously undescribed or untargeted actions of ER. The flow chart outlines one strategy. Of course, a series of experiments that is individualized for each small molecule and pathway is required to identify their site and mechanism of action.
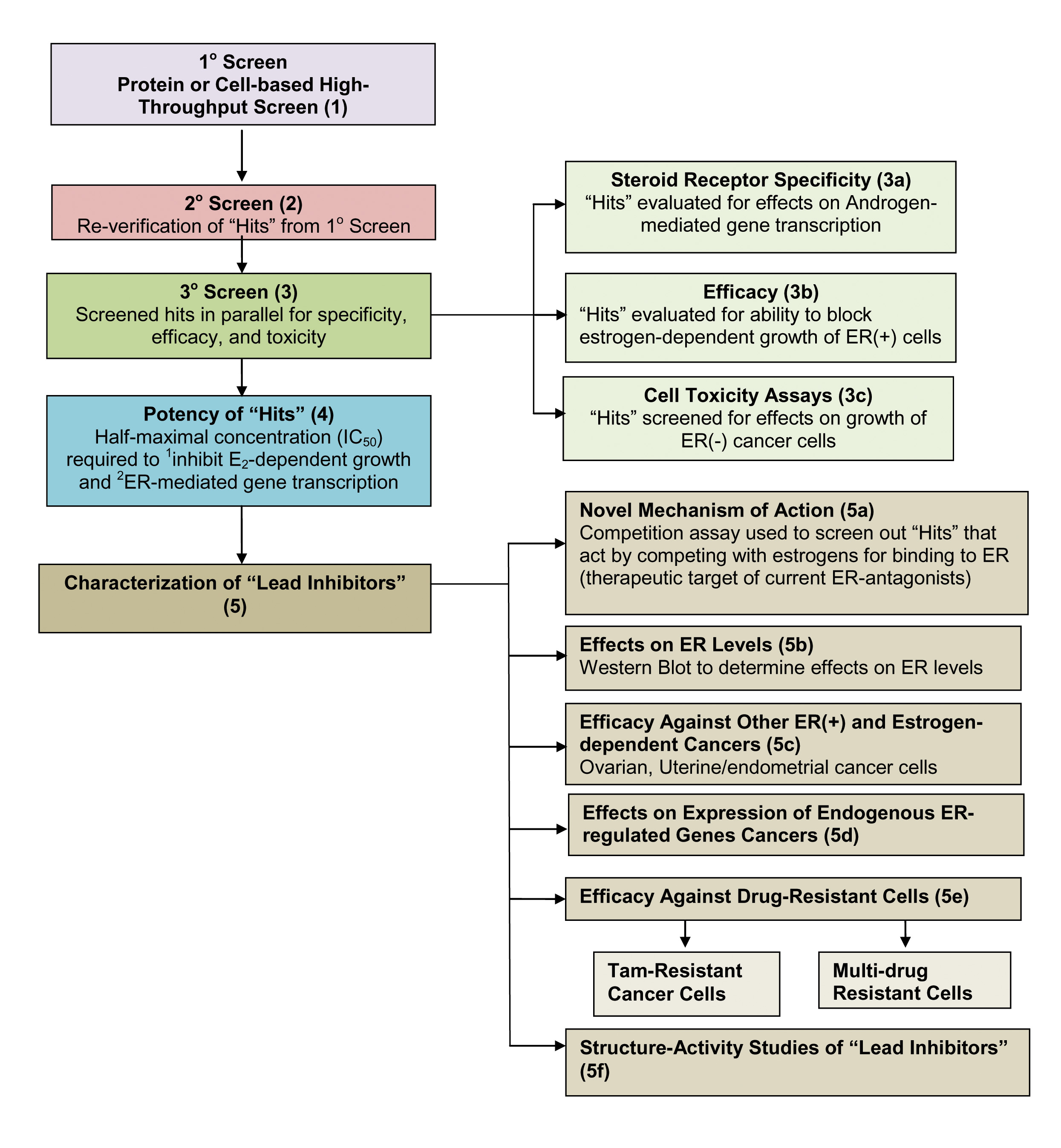
To identify new small molecule inhibitors of pathways important in estrogen-ER induced cancer cell proliferation, we developed a cell-based high throughput screen and then performed an unbiased pathway-directed screen of ~150,000 small molecules to generate a large data set of small molecule biomodulators of estrogen-ERa induced gene expression, followed by evaluating hits for inhibition of estrogen-ER induced cancer cell proliferation and simple follow-on assays to identify a lead inhibitor likely to exhibit a novel mode of action. The results of this currently unpublished work provide the foundation for much of our current research.
Our lead estrogen receptor anticancer drug candidate both induces rapid tumor regression and works by hyperactivating a normal and previously undescribed, pathway of estrogen action in cancer. Graduate students presented 2 talks on this work at the biennial June 2014 International Congress of Endocrinology Four papers describe this ongoing work. One paper is in press in Oncogene, one is in revision and 2 are being written. The abstracts can be found under our publications page.
Our earlier published work is summarized below.
Our small molecule inhibitors were identified using 2 very different types of high throughput screens. To provide a rapid, true equilibrium assay for the interactions of proteins with DNA or RNA recognition sequences we developed the fluorescence anisotropy microplate assay (FAMA) (Wang SY, et al., (2004) Biotechniques, 37:807-817). Here the FAMA is described for analyzing interaction of ER with its fluorescein-labeled DNA recognition sequence, the estrogen response element (ERE). After excitation with polarized light, the relatively small fluorescein-labeled consensus ERE (flERE) undergoes rotational diffusion more rapidly than the time required for light emission. Therefore, the position of the flERE at the time of emission is largely randomized, resulting in depolarization of most of the emitted light (Fig. panel A). When the ER binds to the flERE, the larger size of the flERE-ER complex causes rotational diffusion to be slower, increasing the likelihood that the flERE-ER complex will be in the same plane at the time of emission as it was at the time of excitation. Therefore, the emitted light will be more highly polarized (Fig., panel B). This change is observed as an increase in fluorescence polarization, or the closely-related parameter, fluorescence anisotropy. This version of the assay was used in high throughput screening to identify small molecule inhibitors of the binding of ER to the flERE. Binding of a large coactvator protein, such as SRC1 further increases the size of the complex resulting in the emitted light being highly polarized (Fig., Panel C). We used this method to carry out real-time assays of the binding of full-length coactivators to DNA bound ER (Wang S, et al., (2007) J. Biol. Chem., 282:2765-2775).
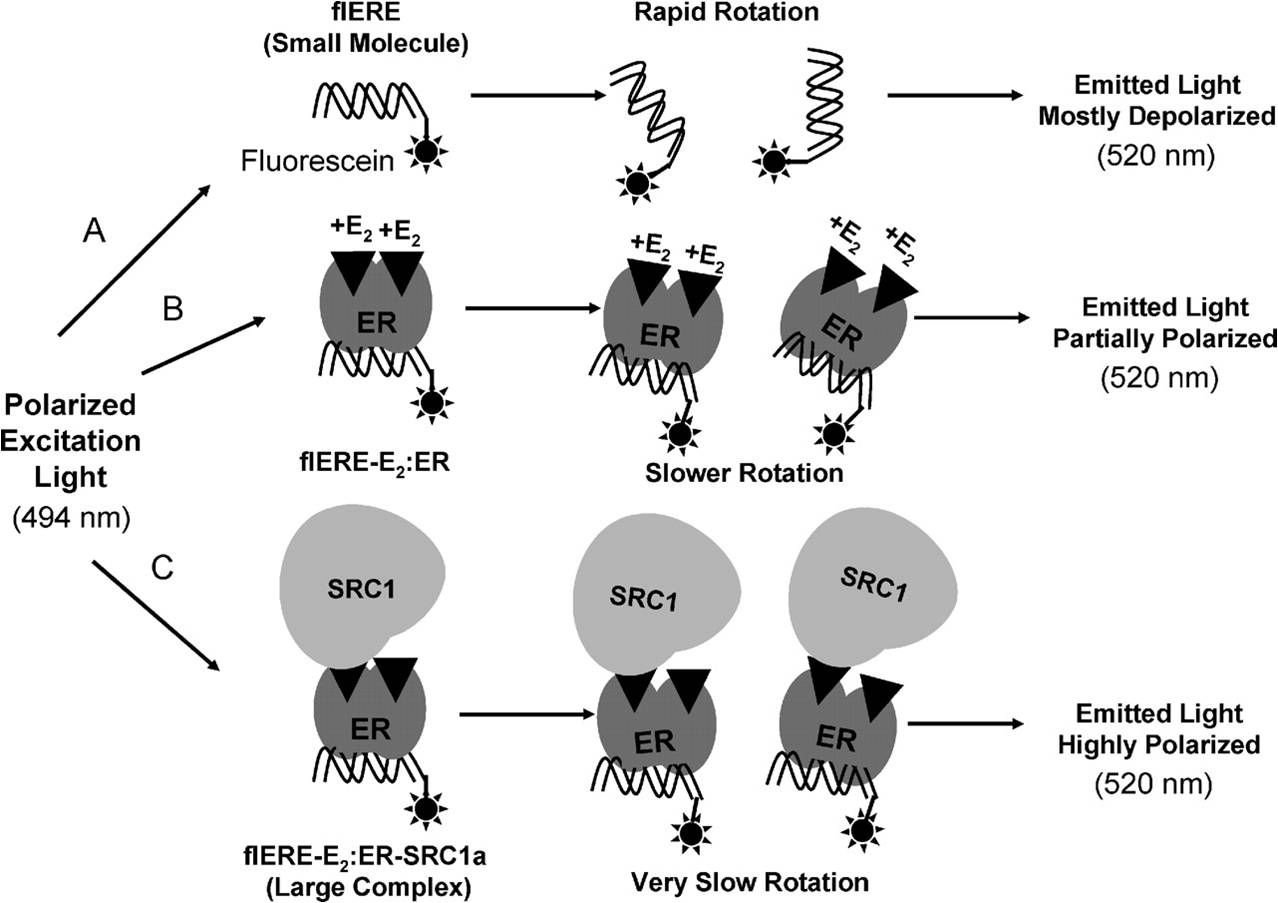
Schematic representation of the fluorescence anisotropy/polarization assay used in screening
and to study coactivator binding. (from Wang et al., 2007)
We used the FAMA to screen for small molecule inhibitors of the binding of ER, PR and AR to their respective fluorescein-labeled DNA response elements. One small molecule theophylline, 8-[(benzylthio)methyl]- (TPBM) (see Fig.) was selected for further study. TPBM selectively inhibits ER, with little or no effect on other steroid receptors and is effective in cells resistant to tamoxifen. TPBM blocked estrogen-dependent growth of cancer cells. Using quantitative RT-PCR to measure the PI-9 mRNA level and quantitative chromatin immunoprecipitation (ChIP) to measure occupancy of the PI-9 estrogen responsive region, we observed a good correlation between the extent to which TPBM inhibits induction of the proteinase inhibitor 9 (PI-9) mRNA and the extent to which TPBM reduces E2-ERα occupancy at the PI-9 gene. Thus, the primary mechanism by which TPBM exerts its intracellular action is by decreasing interaction of ERα with regulatory regions of estrogen-responsive genes. It is likely that interaction of TPBM with ERα induces a conformational change in the receptor, and that one result of this conformational change is decreased association of E2-ERα with EREs. TPBM is a useful research tool to probe the role of DNA binding in ER-mediated processes (for example: Powell et al., (2010) J. Biol. Chem., 285:16125-16134. PMCID: 2871481).
To identify more potent ERα inhibitors, we used a cell-based screen to evaluate ~200 small molecules structurally related to TPBM and identified a much more effective ER inhibitor, butyrophenone, p-fluoro-4-(1,2,3,6,-tetrahydro-1,3-dimethyl-2-oxo-6-thionpurin-8-ylthio) (TPSF) (Fig.). TPSF is ~15 more potent than TPBM. TPSF potently and selectively inhibits expression of ER-regulated genes, inhibits estrogen dependent growth of breast cancer cells and is effective in multiple models for tamoxifen resistant breast cancer. Although TPBM and TPSF are very similar structurally (see Fig.), they have very different effects on ERα. While TPBM inhibits in vitro binding of E2-ERα to a labeled ERE in vitro, TPSF does not. TPSF strongly reduces ERα levels in breast cancer cells, while TPBM has little or no effect on the level of ERα. Since TPSF has very little or no effect on the levels of AR and GR, TPSF is highly selective for down-regulation of ER. Our data indicates that TPSF influences receptor levels at least in part due to its ability to enhance proteasome-dependent degradation of ERα (Kretzer et al., (2010) J. Biol. Chem., 285:41863-41873. PMCID: 3017855).
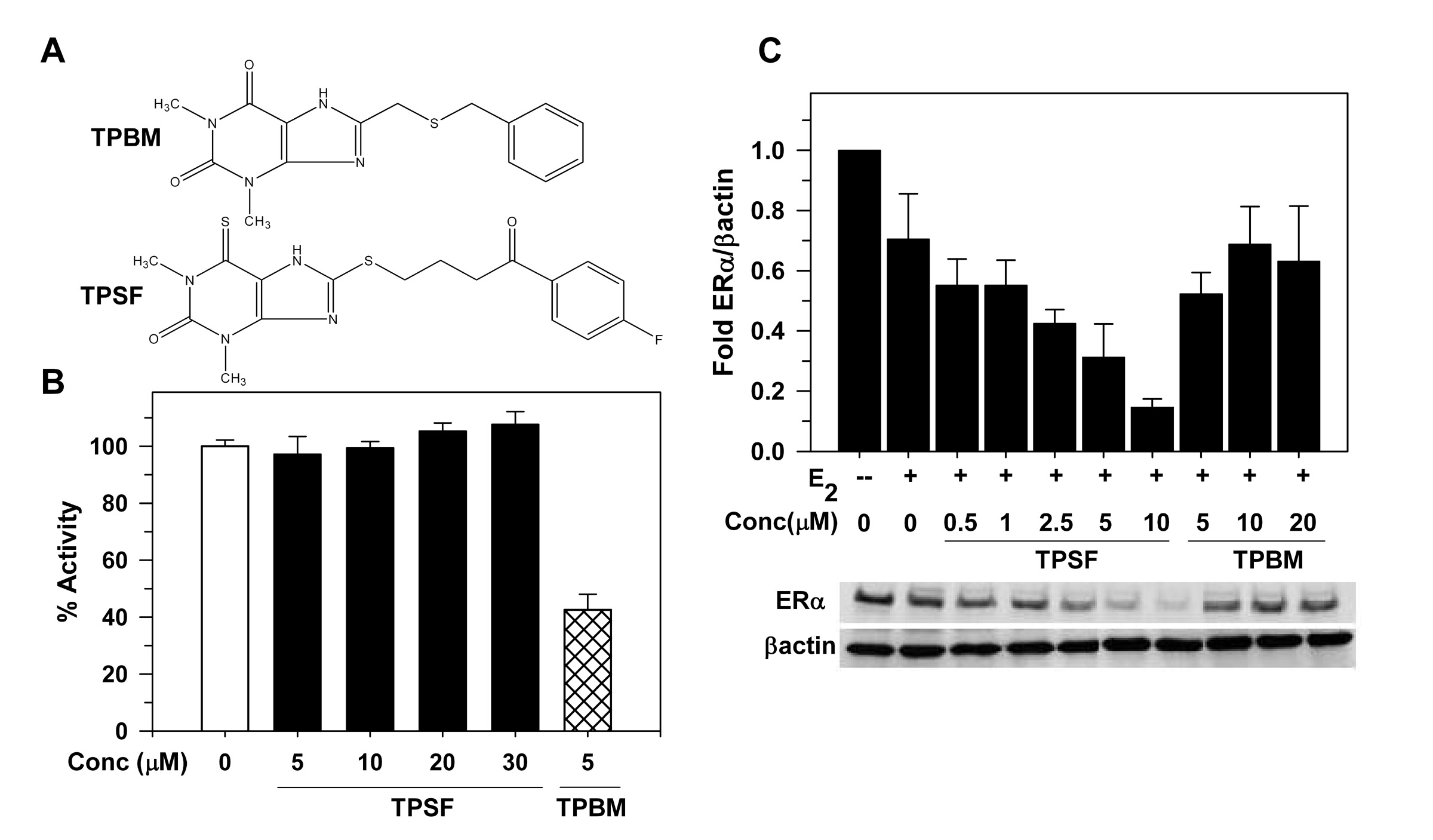
Structurally similar TPBM and TPSF have very different sites of action.
A) Structures of the ER inhibitors, TPBM and TPSF. (B) In contrast to TPBM, TPSF does not inhibit binding
of ER to the flERE. (C) Western blot shows that TPSF, but not TPBM, elicits a concentration-dependent
decline in ER levels. (from Kretzer et al., 2010 and Wang et al., 2011)
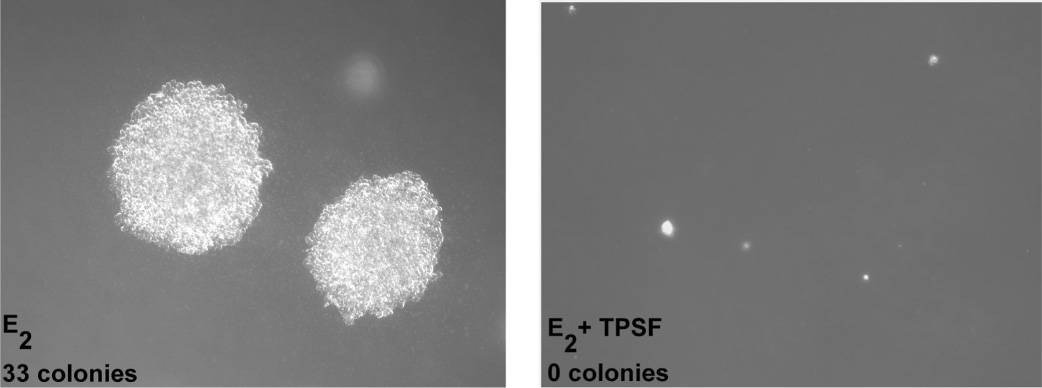
TPSF inhibits growth of MCF-7 cells in soft agar.
How might we account for the different modes of action of these two closely related small molecules?
We hypothesize that just as binding of estrogens and tamoxifen to the same site on ER results in functionally distinct ER conformations, binding of TPBM, and the more potent TPSF, may elicit distinct ER conformations that are associated with different modes of action.
Efforts to understand the actions of TPSF and to identify and characterize novel ER inhibitors are ongoing.
We used stably transfected cell lines epxressing the estrogen (ER), androgen (testosterone) (AR), glucocorticoid (GR) and progesgterone (PR) receptors to test for structural motifs associated with preferential inhibition of PR. We screened approximately 200 small molecules structurally related to TPBM and TPSF for the ability to inhibit hormone-dependent induction of reporter genes activated by each receptor. We identified a structural motif associated with selective inhibition of progesterone receptor. Unlike RU-486/mifepristone and other competitive PR inhibitors that also inhibit GR, the non-competitive PR inhibitors we identified and characterized do not inhibit GR and are useful tools for analyzing the actions of PR and its role in cancer and other diseases.
Selected Publications
Mao C, Pattterson NM, Cherian, MT, Aninye IO, Zhang C, Montoya JB, Cheng J, Putt KS, Hergenrother PJ, Wilson EM, Nardulli AM, Nordeen SK and Shapiro DJ (2008) A new small molecule inhibitor of estrogen receptor α binding to estrogen response elements blocks estrogen-dependent growth of cancer cells. J Biol Chem., 283: 12819-12830. PMCID: 2442351. (Recommended by Faculty of 1000)
Kretzer NM, Cherian M, Mao. C, Aninye I, Reynolds P, Schiff R, Hergenrother PJ, Nordeen SK, Wilson EM and Shapiro DJ (2010) A non-competitive small molecule inhibitor of estrogen-regulated gene expression and breast cancer cell growth that enhances proteasome-dependent degradation of estrogen receptor α. J. Biol. Chem., 285:41863-41873. PMCID: 3017855.
Shapiro, DJ, Mao C and Cherian MT (2011) Small molecule inhibitors as probes for estrogen and androgen receptor action. J. Biol. Chem. 286:4043-4048. PMCID: 3039394
Andruska N, Mao C, Cherian M, Zhang C and Shapiro DJ (2012) Evaluation of a luciferase-based reporter assay as a screen for inhibitors of estrogen-ERα-induced proliferation of breast cancer cells. J. Biomolecular Screening, 17:921-932. PMID 22498909.
Aninye IO, Berg KC, Mollo AR, Nordeen SK, Wilson, EM, and Shapiro DJ (2012) 8-Alkylthio-6-thio-substitited theophyllene analogues as selective noncompetitive progesterone receptor antagonists, Steroids, 77: 596-601. PMID 22421057.
Zhang C, Nordeen SK and Shapiro DJ (2013) Fluorescence anisotropy microplate assay to investigate the interaction of full-length steroid receptor coactivator-1a with steroid receptors Methods Mol. Biol. 977:339-351. PMID: 23436375.
Andruska N, Zheng X, Yang X, Helferich WG and Shapiro DJ (2015) Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor α-positive breast cancer. Oncogene, 34(29):3760-9. PMCID: 4377305.
Andruska N, Zheng X, Yang X, Mao C, Cherian MM, Mahapatra L, Helferich WG and Shapiro DJ (2015) Estrogen receptor α inhibitor activates the unfolded protein response, blocks protein synthesis, and induces tumor regression. PNAS, 112(15): 4737-42. PMCID: 4403155. (Highlighted Paper)
Zheng X, Andruska N, Yu L, Mao C, Kim JE, Livezey M, Helferich WG and Shapiro DJ (2016) Interplay between steroid hormone activation of the unfolded protein response and nuclear receptor action. Steroids, 114:2-6. PMCID: 5035163.
Shapiro DJ, Livezey M, Yu L, Zheng X and Andruska N (2016) Anticipatory UPR Activation: A Protective Pathway and Target in Cancer. Trends in Endocrinology and Metabolism, 27(10): 731-41. PMCID: 5035594.
Zheng X, Andruska N, Lambrecht MJ, He S, Parissenti A, Hergenrother PJ, Nelson ER and Shapiro DJ (2016) Targeting multidrug-resistant ovarian cancer through estrogen receptor α dependent ATP depletion caused by hyperactivation of the unfolded protein response. Oncotarget, Epub. PMID: 27463013.
Mao C, Livezey M, Kim JE and Shapiro DJ (2016) Antiestrogen Resistant Cell Lines Expressing Estrogen Receptor α Mutations Upregulate the Unfolded Protein Response and are Killed by BHPI. Scientific Reports, 6:34753. PMCID: 5054422.