Milestones in
Photosynthesis Research
In: "Probing Photosynthesis: Mechanisms, Regulation And Adaptation", edited by Mohammad Yunus, Uday Pathre, and Prasanna Mohanty, published in 2000by Taylor & Francis, London, pp.9-39. A pdf file of the chapter and a correct Figure 3 is available by sending an E-mail to gov@uiuc.edu
GOVINDJEE
Departments of Biochemistry, Plant Biology and Center of
Biophysics and Computational Biology
University of Illinois at Urbana-Champaign
TABLE OF CONTENTS
PHOTOSYNTHETIC UNIT: ANTENNA AND REACTION CENTRES
THE TWO PIGMENT SYSTEMS AND THE TWO LIGHT REACTIONS
Electron Transport: Acceptor Side of Photosystem II
The task of writing a chapter on milestones in photosynthesis research is difficult. because there are so many milestones that I may not be able to do justice to them all. Thus, at the very outset, I beg forgiveness for incompleteness and I urge the readers that they should not consider this chapter as a record of the history of photosynthesis. Further, this chapter will not present milestones (including the ones marking one tenths of a mile) in a linear chronological manner, but according to specified topics. For an earlier thought-provoking article on the conceptual development in photosynthesis, see Myers (1974), for historical development, see Huzisige and Ke (1993), and for an introductory overview on photosynthesis, see Whitmarsh and Govindjee (1995). An overview on the milestones in the area of chlorophyll a fluorescence has been presented earlier (see Govindjee, 1995; also see Govindjee et al., 1986, and the chapter by Strasser and coauthors, this volume); thus, it will not be covered here unless I consider it pertinent to my discussion.
A tribute. This chapter is written in honor of my dear friend, a trusted colleague and a World leader in the field of Photosynthesis, Dr. Prafullachandra Vishnu Sane. I refer here only to three of his contributions. A thought-provoking new model of the distribution of the two photosystems in the thylakoid membrane system was presented elegantly by him in collaboration with Rod Park in their classic review (Park & Sane, 1971; also see Sane, 1977). His pioneering research, in collaboration with V.G. Tatake & T.S. Desai, in devising methods to record the most highly resolved thermoluminescence bands from the photosynthetic material, and for assigning these bands not only to photosystem II, but also photosystem I, have been reviewed by him in a chapter in a book (see Sane & Rutherford, 1986). I would also like to mention his discovery of heat-induced "state changes" in photosynthetic systems (see Sane et al., 1984).
The primary source of energy for nearly all life on Earth is the Sun. As early as 1845, Robert Mayer, who provided us with the Law of Conservation of Energy, had already recognized that plants convert light energy into chemical energy on a massive scale. Photosynthesis is the physico-chemical process by which oxygenic (plants, algae, cyanobacteria and prochlorophytes) and anoxygenic (photosynthetic bacteria) organisms convert light energy into redox chemical energy on a global scale. Each year 4x1018 kilojoules of free energy is stored in reduced Carbon by this process. In terms of carbon, each year about 1011 metric tons of CO2 are converted into organic matter by photosynthesis. According to Woese et al. (1990) the living organisms can be divided into three groups: archea; bacteria; and eukarya. Archea do not engage in true photosynthesis although a bacteriorhodopsin-containing organism Halobacterium salinarium (formerly H. halobium) can convert light energy into adenosine tri-phosphate (ATP). Photosynthetic bacteria and cyanobacteria, mentioned above, are clearly bacteria as the name implies, whereas plants and algae are eukarya. Oxygenic photosynthesis provides us with both food and oxygen, and anoxygenic photosynthesis only with food, needed for the survival of almost all living organisms except certain bacteria (two examples are Methanococccus janaschii and Methanobacterium autotrophicum). In addition, ancient photosynthesis is still providing us with fossil fuel; at the rate we are using it, it is not going to last forever. We need to understand the basics and the historical development of photosynthesis since it is the only process that can provide us with food, fuel and oxygen needed for the ever-increasing population, and since it is the only major process that may utilize the global increases in CO2 we are experiencing. It is now obvious that by the year 3,000, we expect the World's population to be 13 billion, but at the same time the available land for food production/person is decreasing at an alarming rate caused by our follies (increased deforestation and fossil-fuel burning) (Kendall et al., 1997; Sinha & Swaminathan, 1991; Swaminathan, 1998). Drastic measures are required to overcome this impending crisis that is already on our doorstep!
Within eight minutes of its origin, sunlight reaches the
photosynthetic organisms on the Earth; almost billion chlorophyll
molecules in a single chloroplast function to capture this energy
within femtoseconds (10-15 s) (Note just for fun: there are as
many femtoseconds in a second as there are seconds in 31 million
years.) In what follows, I present a historical and a conceptual
perspective on the milestones and breakthroughs in photosynthesis
research dealing with the conversion of light energy into
chemical energy in the form of reduced nicotinamide adenine
dinucleotide phosphate (NADPH) and adenosine tri phosphate (ATP),
which leads to the production of food from CO2 (see a general
review by Whitmarsh & Govindjee, 1995). The steps for the
latter were first fully deciphered by M.Calvin, A. Benson and J.
Bassham at the University of California at Berkeley, for which
Calvin was rewarded with a Nobel Prize in Chemistry in 1961. This
aspect of photosynthesis will not be included in my chapter (see
e.g., Bassham et al., 1954; Calvin, 1989 for the Calvin cycle;
and Cleland et al., 1998, for the current understanding of the
mechanism of the function of its key enzyme RUBISCO). Later,
through the work of Kortsckak et al. (1965), Karpilov (1960) and
Hatch & Slack (1966), it was discovered that in certain
plants (such as sugarcane), the first product of CO2 fixation is
a 4-C organic acid (e.g., oxaloacetic acid) in contrast to what
Calvin and co-workers had found in the green alga Chlorella
where the first stable product of CO2 fixation was a 3-C
organic acid phosphoglyceric acid. This C-4 pathway is an
appendage to the Calvin cycle (C-3 pathway) in plants such as
sugarcane and pineapple. The discovery of the C-4 pathway has
been elegantly presented by Hatch (1992). An important
consideration about photosynthesis must be mentioned here: almost
complete photosynthesis, i.e., CO2 fixation, occurs within the
chloroplast as first convincingly shown by Arnon (1955).
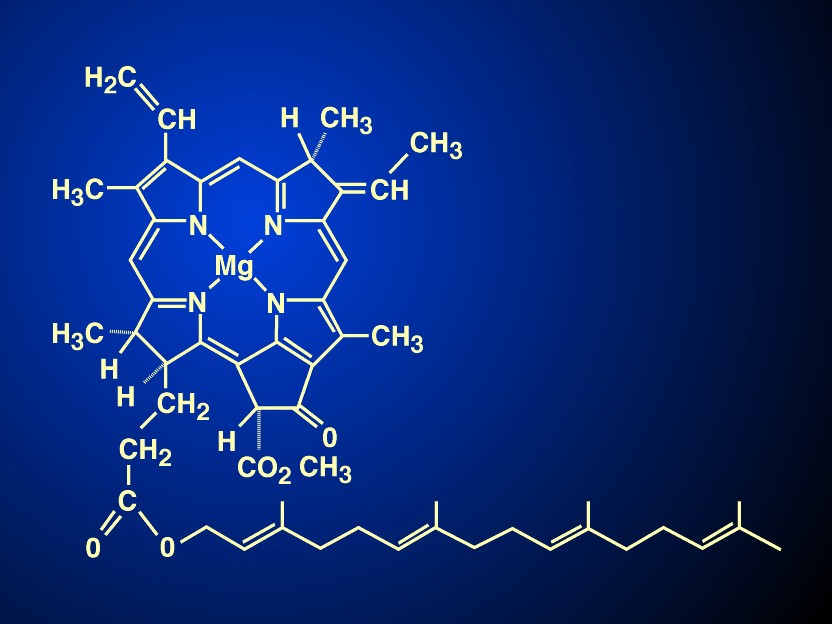
The most visible colour on our Earth is green, a colour quite pleasing to our eyes. Nature evolved chlorophylls to capture sunlight; their structures were such that they could absorb blue and red light, and transmit green light giving leaves the colour green; nature also evolved rhodopsin in our eyes for us to see efficiently this green light. A major landmark in photosynthesis research was the elucidation of the physical and chemical properties of the chlorophyll molecule, a tetrapyrrole that has conjugated (alternating) single and double bonds giving it the spectroscopic properties of absorbing blue and red light, and a unique chemical structure allowing it to have different binding properties to different set of amino acids in various proteins to make it into light absorbing antenna, as well as reaction center chlorophylls labelled as P680 or as P700 (P for pigment trap, the numbers indicating the wavelength maxima of the first singlet excited state, in nanometers). Only P680+/P680 was destined to have the redox potential so positive (Em,7 :~ +1.1 V) that it can oxidize water to molecular oxygen (average Em,7 +0.8 V), whereas P700+/P700 (Em,7 +0.4 V) is only able to oxidize intermediates such as the hemes in cytochromes or copper in plastocyanin.
The elucidation of the chemical properties and the structure of chlorophylls was rewarded with the Nobel prizes in Chemistry to Richard Wilstätter (in 1915), to Hans Fischer (in 1930), whereas its total synthesis was recognized with a Nobel prize, also in Chemistry, to R. Woodward (in 1965). It was Duysens (1952) who had first used the term "P" for pigment representing a few reaction centre chlorophyll a or bacteriochlorophyll molecules in his Ph D thesis at the State University of Utrecht. The term P870 for bacterial reaction centre was then coined. Kok (1956, 1957), who was then at the Wageningen Agriculture University's 'ship-shaped' building, discovered P700, the reaction centre chlorophyll of what we now call Photosystem I (PSI, see a later section), whereas it was H.T. Witt and coworkers in Berlin (see Döring et al., 1969) who discovered P680, the reaction centre chlorophyll of what we now call Photosystem II, PSII. I note that my mentor and second advisor E. Rabinowitch had long suspected its presence, and Rabinowitch & Govindjee (1965) had named it as such prior to its discovery.
Although P680 and P700 are "special pairs" of chlorophyll a molecules, they are entirely different mainly because of their binding to different but specific amino acids in PSII and PSI reaction centre proteins. There are other chlorophylls too: Chl b, Chl c, and Chl d. In spite of my first advisor R. Emerson's early speculative and tentative ideas, Chl b and Chl c are only antenna pigments, i.e., they function only as light-harvesters in plants (as well as in green algae & prochlorophytes) and brown algae (as well as in diatoms & dinoflagellates), respectively. The recognition of Chl d as the major antenna pigment in Acaryochloris marina, a prochloron like prokaryote was discovered by Miyashita et al.
(1996). Whether Chl d can serve as the reaction centre Chl is under investigation in Japan. In addition, the possible role of Zinc chlorophylls versus Magnesium chlorophylls is also being actively investigated by S. Itoh and coworkers (personal communication). If confirmed, the latter two would break the stereotypic knowledge we had thus far regarding the uniqueness of Chl a as the only reaction centre chlorophyll of oxygenic photosynthesis.
The importance of the chlorophylls has been recognized by at least two books, edited by Vernon & Seely (1966) and by Scheer (1991). And finally, the recent discovery of a new type of bacteriochlorophyll, labelled as bacteriochlorophyll g (BChl g) (see Gest & Favinger, 1983; Brockman & Lipinski, 1983; Figure 1; also see Amesz, 1996) may have completed the discovery of all the various types of chlorophylls present in nature. BChl g resembles BChl b in having an ethylidene group on C8, which in the presence of oxygen and light, isomerises to give a vinyl group. Further, BChl g has a second vinyl group on C3; the product of its isomerisation is a molecule which is very similar to Chl a, the pigment of plants and green algae. Thus, heliobacteria acquire a possibly new evolutionary significance.
The key character of Chl a is that its bound forms, P680* and P700*, are photoenzymes converting light into chemical redox energy within a few picoseconds, acting as the world's most efficient solar battery:
P680 + Pheophytin + photon (or exciton)-----> P680+ + Pheophytin- (1)
(uphill electron transfer overcoming about 1.7 electron volts of energy barrier by a red photon)
P700 + Ao + photon (or exciton)-----> P700+ + Ao- (2)
(uphill electron transfer overcoming about 1.1 electron volts
of energy barrier by a far-red photon; Ao is a chlorophyll
monomer bound at a specific site in Photosystem I)
PHOTOSYNTHETIC
UNIT: ANTENNA AND REACTION CENTRES
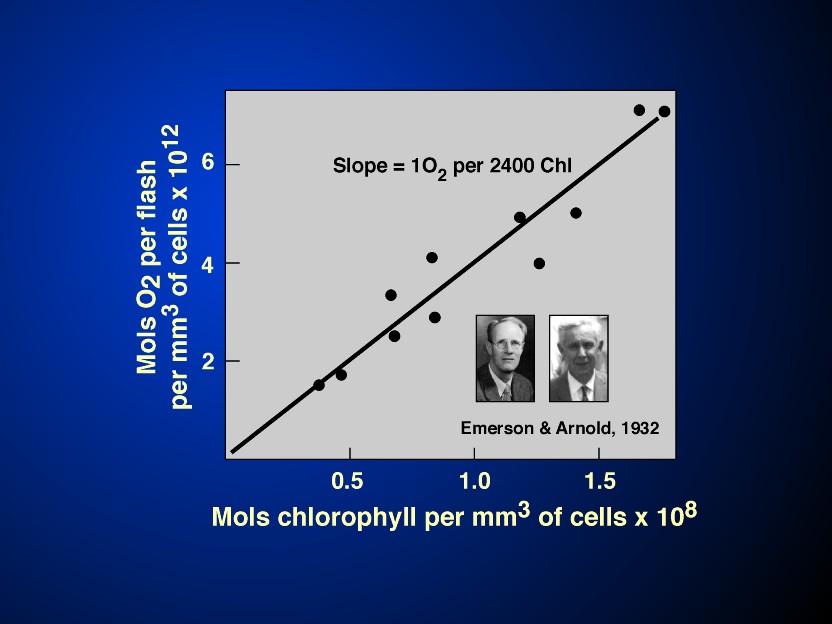
The terms antenna and the reaction centres have already been alluded to above. The concept is simple: a large number of light harvesting molecules function to capture light energy and act as if they are "antennae"; this captured light energy, in the form of excitation energy (or excitons) is transferred to a few pigment molecules that serve as reaction centre pigment molecules where primary photochemistry takes place. By primary photochemistry, we mean conversion of light (or excitation) energy into redox chemical energy that is then stored to do useful work. The birth of this concept, i.e., of a photosynthetic unit, that includes the two components, the antenna and the reaction centre, took place in 1932 in the Kirchhoff Laboratory of Biological Sciences, at the California Institute of Technology in Pasadena, CA. There were two players: assistant Professor of Biophysics Robert Emerson (see Rabinowitch, 1961) and an undergraduate student William Arnold. Emerson & Arnold (1932a,b) discovered that under the most optimal condition of photosynthesis (single turn-over brief and saturating flashes of light, with optimal dark times between them), a maximum of only one oxygen molecule was evolved per about 2,400 chlorophyll molecules present in the green alga Chlorella (Figure 2) although the maximum quantum yield of oxygen evolution (i.e., the number of O2 evolved per quantum absorbed) must have been very high, 1/8 to 1/10 in today's numbers). Thus, the existence of a Unit (and a photoenzyme) was suggested. It was, however, Gaffron & Wohl (1936) who provided the correct and complete interpretation of the highly elegant and sophisticated experiments of Emerson & Arnold: light absorbed by most of the chlorophyll molecules in the "photosynthetic unit" is transferred to the "photoenzyme" for chemistry. The 1932 experiments were unique in another sense: they were the first experiments in Science to use repetitive flash technique increasing the Signal/Noise ratio allowing precise measurements of small quantitities of oxygen evolution by manometric methods. It is of historical interest to mention here that both the techniques of manometry and the green alga Chlorella, used in this work, were introduced to Science by Emerson's own Professor Otto Warburg, who had received the Nobel prize in Physiology & Medicine in 1931 for his studies on biochemistry. The 1932 papers of Emerson & Arnold are now classical papers and continue to be regularly cited even after 66 years of their publication. This milestone discovery was recently discussed in an elegant paper by Myers (1994), in a special issue of "Photosynthesis Research", edited by Govindjee et al. (1996), and in a recent book by Wild & Ball (1997). The Emerson-Arnold "Photosynthetic Unit" has a functional definition : about 2,400 chlorophyll molecules cooperate to evolve one oxygen molecule (and, thus, reduce one CO2 molecule). As noted above, photosynthetic unit includes antenna complexes (the light harvesters) and the reaction centres. Exciton migration occurs in femto-to- picosecond range, among the photosynthetic pigments located in the protein complexes, the antenna proteins (see Hoff & Amesz, 1995; Hoff & Deisenhofer, 1997). The first kinetic evidence for the excitation energy transfer was obtained from time-resolved ultrafast fluorescence spectroscopy experiments initiated by Brody & Rabinowitch (1957) at UIUC, Urbana, Illinois. A wonderful example of this was shown by Yamazaki et al.(1984) in red algae where one can literally watch the excitons move from phycoerythrin to phycocyanin, then to allophycocyanin, and finally to chlorophyll a in a cascade-like manner.
Major breakthroughs in the understanding of the antenna structure at the atomic level have been through the availibility of X-ray and electron diffraction crystallography. Kühlbrandt et al.(1994) have provided the atomic level structure of the largest antenna of Photosystem II, the Light-harvesting complex II b; here one can see where the Chl a and Chl b molecules are anchored. On the other hand, McDermott et al. (1995) and Walz and Ghosh (1997) have provided the atomic level structures of two light-harvesting antenna complexes of anoxygenic photosynthetic bacteria; and, Hofmann et al. (1996) have provided the atomic level structure of the peridinin-chlorophyll protein complex of the dinoflagellate Amphidinium carterae. These provide the most fascinating view of the grand design of the architecture of the photosynthetic apparatus responsible for the capture and trapping of the excitation energy. These newer structures had been preceded by the structure of the chlorobium protein complex (the so-called Fenna-Mathews-Olson protein, see Fenna & Mathews, 1976; Mathews & Fenna, 1980; Tronrud et al., 1986), of phycoerythrin (Fiener & Huber, 1993) and of phycocyanin (Schirmer et al., 1986). Thus, we are now able to ask meaningful questions about the detailed physico-chemical reactions in the antenna and finally begin to understand the molecular mechanism of excitation energy transfer in photosynthesis, an area that had been dominated in the past mainly by speculative and theoretical arguments.
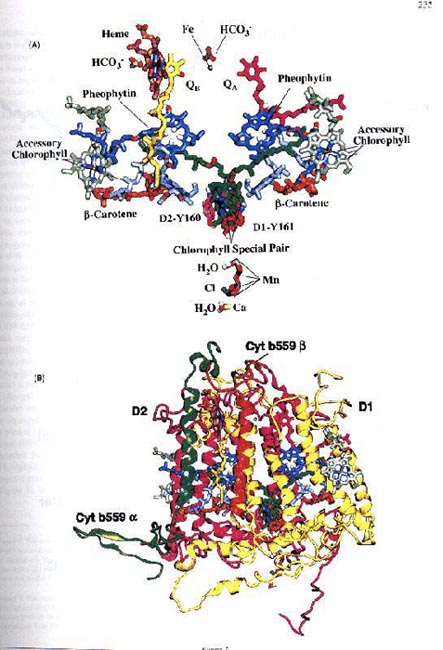
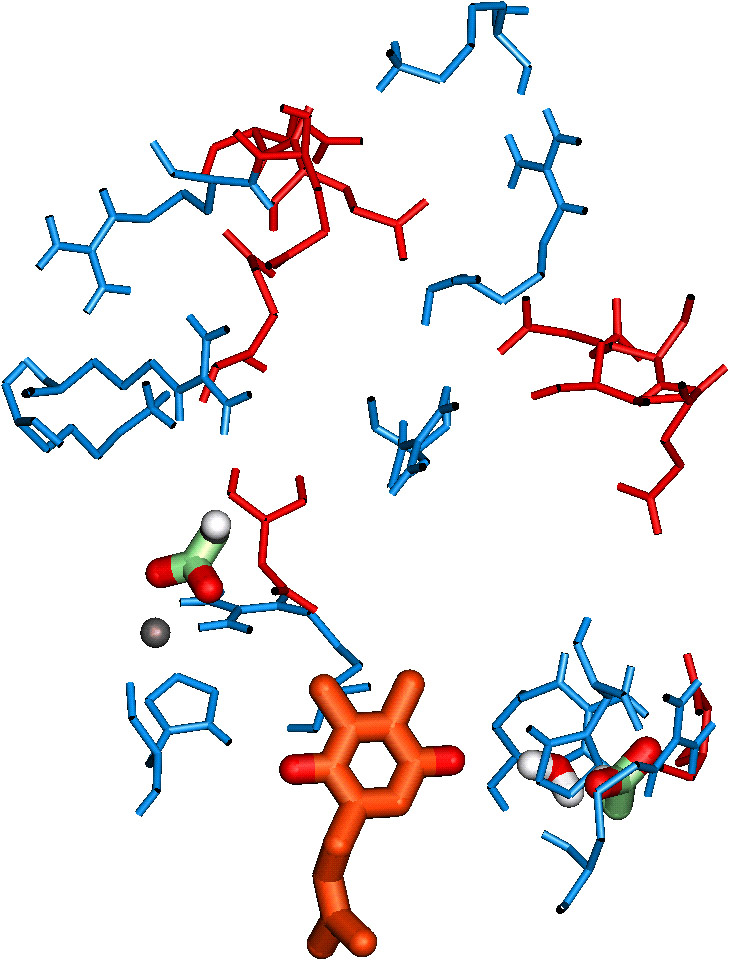
The conversion of light energy into chemical energy occurs in the World's most efficient solar battery, in the picosecond time range, in the reaction centre molecules. This conversion leads to the formation of the primary charge-separated state, P+A-, from PA, where P is the special reaction centre molecule, as already mentioned, and A is the primary electron acceptor. Examples of this reaction were already given under the section CHLOROPHYLLS. This is the only true light reaction of photosynthesis; all others can, in principle, occur in darkness. They are the only ones that are "uphill" because the free energy is positive due to the unfavourable difference in redox potentials of P/P+ and A/A-. These primary reactants and those involved in charge stabilization are located in the reaction centre complexes. The X-ray diffraction crystal structure of the reaction centre complex of the anoxygenic photosynthetic bacterium Rhodopsuedomonas viridis was the first one to be published, and was rewarded with the 1988 Nobel prize in Chemistry to H. Michel, J. Deisenhofer and R. Huber (see Deisenhofer et al., 1984,1985, 1995; Feher et al., 1989; Feher, 1998). Recently, Rhee et al. (1997) have provided a crude structure of P680-containing Photosystem II reaction centre complex at 8 Å; there are no details available. However, knowledge-based atomic level models of Photosystem II are now available (see e.g., Xiong et al., 1996, 1998; see Figure 3 and its legend ; also see a partial model by Coleman et al., 1997). Just as femtosecond-to-picosecond absorption spectroscopy have provided information on the primary charge separation in anoxygenic photosynthetic reaction centre (see e.g., Hoff & Deisenhofer, 1997; Zinth et al., 1998), information on Photosystem II are also now available (see Greenfield and Wasielewski, 1996; and Groot et al., 1997). Greenfield et al. (1997) have suggested that at ambient temperatures two time constants of charge separation can be measured:(1) approximately 8 ps, an upper limit for charge separation, due in part to equilibration of excitons among the core reaction centre chlorophylls (the "red" pool, R); and (2) approximately 50 ps, due, in part to equilibration of excitons among the accessory chlorophylls (the "blue" pool, B).
Chl a (B)* Chl a (R) P680 Pheo 50 ps >Chl a (B) Chl a (R)* P680 Pheo (3)
Chl a (B) Chl a (R)* P680 Pheo 8ps >Chl a (B) Chl a (R) P680* Pheo (4)
Chl a (B) Chl a (R) P680* Pheo 3ps >Chl a (B) Chl a (R) P680+ Pheo- (5)
We believe that the actual charge separation time is closer to 3 ps, the same as in anoxygenic photosynthetic bacteria. (In the equations shown above, * represents the molecule with an exciton or a photon.) All of the experiments on the primary photochemistry of photosystem II have been possible because of the success of Nanba & Satoh (1987) in isolating the simplest, although incomplete, reaction centre protein of photosystem II.
In contrast to PSII, a 6 Å resolution structure of
P700-containing Photosystem I (PSI) reaction centre was published
already five years ago by the research group of H.T. Witt of
Berlin (Krauss et al., 1993); now, a 4 Å resolution structure of
the same complex is available (Krauss et al., 1996). In addition
to differences in the details, e.g., PSI being a Fe-S containing
reaction centre, the PSI reaction centre complex contains also
the core antenna pigment as well as the antenna chlorophylls. I
consider this work to be one of the major milestones in the
history of photosynthesis.
THE TWO PIGMENT
SYSTEMS AND THE TWO LIGHT REACTIONS
Electron transfers occur in pico-to-millisecond time range;
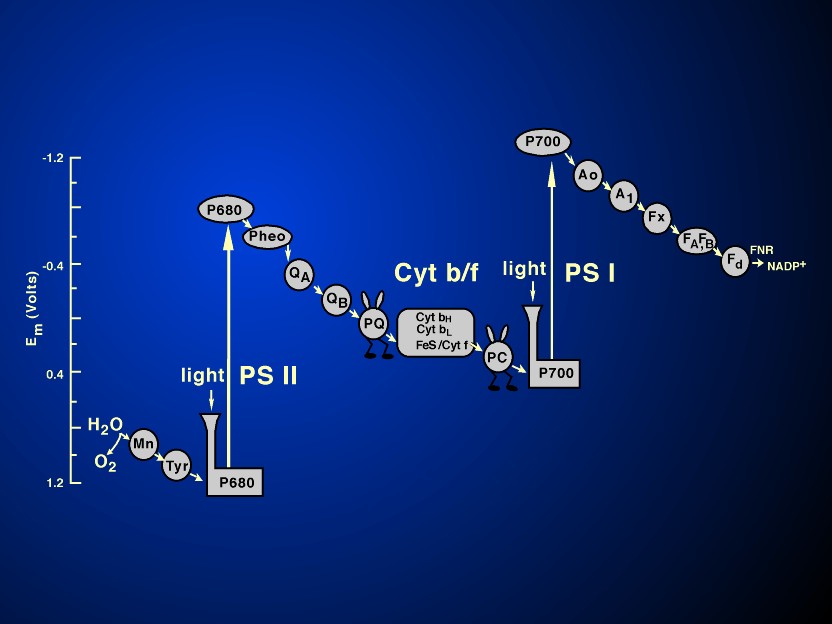
they involve, in oxygenic photosynthesis, three major protein
complexes: Photosystem II (water-plastoquinone oxido-reductase);
Cytochrome b6/f complex (plastoquinol-plastocyanin
oxido-reductase); and Photosystem I (plastocyanin-ferredoxin
oxido-reductase). A fourth ATP synthase is required for ATP
synthesis (see Figure 4 and its legend).
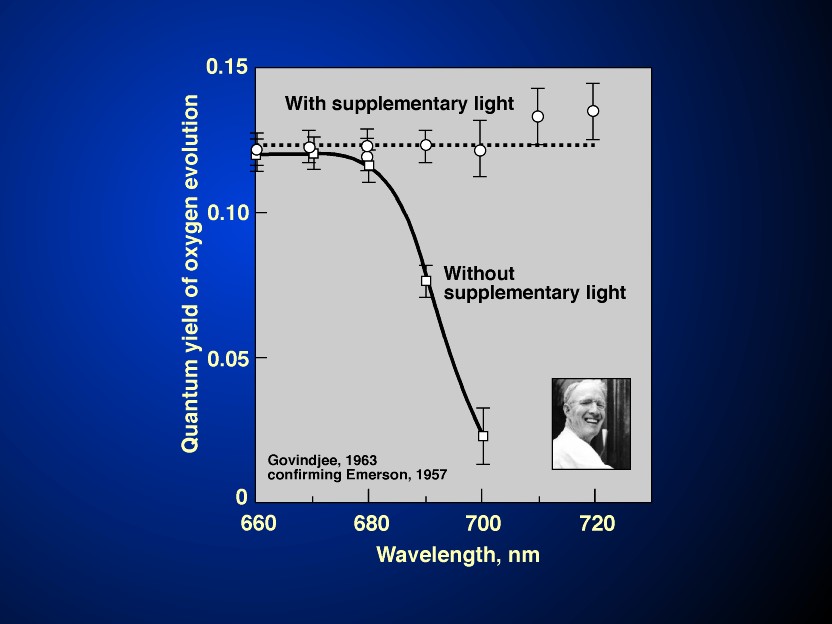
An enigma, discovered by Emerson & Lewis (1943), was that the
quantum yield of oxygen evolution decline suddenly beyond 680 nm,
when chlorophyll a was still absorbing light; this
so-called "red-drop" phenomenon led to the current
concept of the two light reactions and two pigment systems when
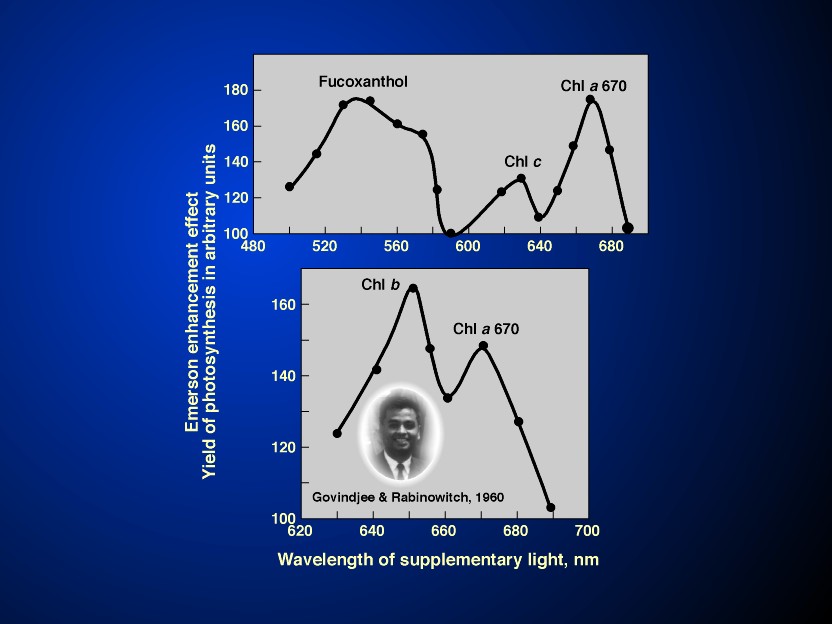
Emerson et al. (1957) discovered the Enhancement effect in
photosynthesis. This concept is based on solid grounds: from the
early ideas of E. Rabinowitch (1945, p.162; 1956, p. 1862) and of
Hill & Bendall (1960). In the experiments on the so-called
Emerson Enhancement in photosynthesis (see Figures
5 & 6 and their legends), or in
the Hill Reaction, yield in far-red light (light I) by
simultaneous exposure to shorter wavelength light (light II), is
enhanced (Emerson et al., 1957; also see Emerson, 1958;
Emerson & Rabinowitch, 1960; Govindjee & Rabinowitch,
1960; R. Govindjee et al., 1961, 1962, 1964; and a review by
Myers, 1971). Further, the crucial experiments on the
antagonistic effect of light I and II on the redox state of P700
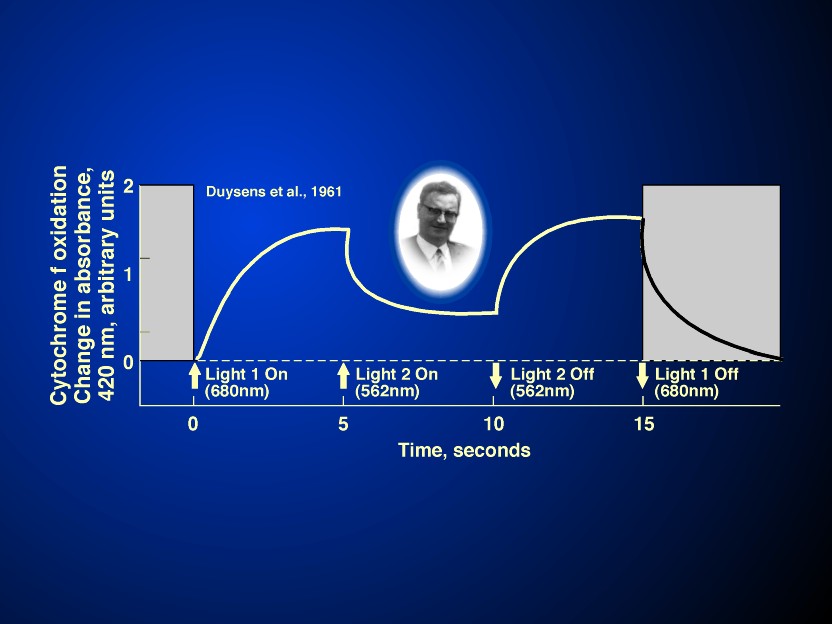
(Kok, 1959; see Figure 7 and its legend),
on the Chlorophyll a fluorescence yield (Govindjee et al., 1960;
Butler, 1962; Duysens & Sweers, 1963), and, most
importantly on the redox state of cytochrome f (Duysens et al.,
1961; see Duysens, 1989) established the two light reaction
scheme of photosynthesis. In fact, the existence of the series
scheme of photosynthesis is best proven by light I (absorbed in
photosystem I) oxidising cytochrome f and light II (absorbed in
photosystem II) reducing the oxidised cytochrome f (see Figure 8 and its legend). However, without
the fast absorption spectroscopic work on the new intermediate
X-320 (now known to be equivalent to the so-called QA, the first
bound plastoquinone molecule), the history of the discovery of
the two light reactions would be incomplete (see Witt et al.,
1961; Witt, 1991). Figure 9 shows the
current model of the so-called Z scheme of electron transfer in
oxygenic photosynthesis, whereas the legend of Fig.9 describes
the current scheme. (The reader should refer to Figure 4 for the
location of the components described in this figure.)
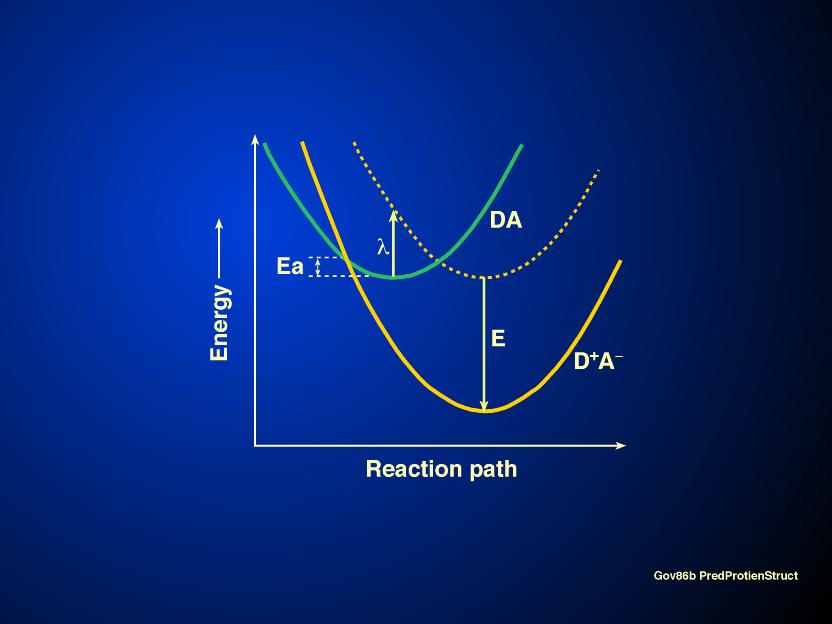
The mechanism by which electrons are transferred from one molecule to another has now been explained by R. Marcus (1993) who was awarded the 1992 Nobel prize in Chemistry for it.. A brief discussion follows. When a system such as e.g., DA is converted into D+A-, i.e., an electron is transferred from D to A, the energy curves of the two states DA and D+A- are shifted with respect to each other on axis of the reaction path (see Figure 10). Marcus defined a new term called the reorganizational energy (l ) by the length of the vertical line from the energy curve of the DA state to where the line hits the D+A- curve. The rate constant of the reaction is given by:
k= A exp ( -D G*/kBT) (6),
where A depends on the nature of the electron transfer reaction, kB is the Boltzmann constant, T is the absolute temperature in Kelvin, and D G* is the activation free energy for the reaction. And,
D G* = l /4 (1+ D Go/l )2 (7),
where D Go is the standard free energy for the reaction, and l
is the reorganizational energy, as mentioned above. A major
prediction of the Marcus equation (7) is that as D Go is varied from zero to some
negative value, D G* decreases and
becomes zero at D Go = -l , then increases when D
Go is made still more negative , i.e., -D Go > l
. The initial decrease of D G* with
increasingly negative D Go
was as expected, but what Marcus equation explained was the rate
of electron flow in what he had called the "inverted
region": increases in D G* when D Go was made increasingly
negative. This prediction was made in 1960, but was confirmed
experimentally only in 1984 by G.L. Closs and coworkers. Marcus
(1993) has extended his theory to explain the characterestics of
electron transfer in the bacterial reaction center. It is
interesting to point out that electron transfers during the
primary charge separation, discussed earlier, do not slow down at
cryogenic temperatures; in fact, they become temperature
independent. Such a phenomenon had been discovered by DeVault
& Chance (1966) (also see DeVault, 1989): it was suggested
that electrons can "tunnel" through barriers as if they
squeeze through with certain probability. Further, if the
energetics and other entropic parameters are constant, electron
transfer rates can be predicted from the distances of the
acceptor from the donor molecules. Moser et al. (1992) showed
that if the acceptor is 1.4 Å further away, the transfer rate
decreases by an order of magnitude. This, known as the
"Dutton ruler", does not explain why electrons flow
only on one side of the bacterial reaction centre molecule, not
the other. Future prospects of understanding the mechanism of
electron transfer are increasing as we begin to have atomic
structures of the various intermediates. For example, we now have
access to atomic level structures of the hydrophilic parts of
cytochrome f (Martinez et al., 1994); of plastocyanin (Redinbo et
al., 1993); and of the Cyt b/c complex of beef heart
mitochondria (Xia et al., 1997) that may have similarities to Cyt
b6/f complex of plants. Zhang et al. (1998) have described a
model of electron transfer in which movement of domains occurs in
cytochrome b/c1 complex; I expect it to have application to
electron transfer in cytb6/f complex. However, in spite of the
available structure of the reaction centre of anoxygenic
photosynthetic bacteria, which have tremendous similarities in
their two halves, we still do not fully understand why the
electrons flow mostly on one, rather than the other side of the
molecule. Here, a combination of molecular biology and physics
& physical chemistry have begun to play important roles in
the understanding of this process. We are still waiting for the
final understanding.
Electron
Transport: Acceptor Side of Photosystem II
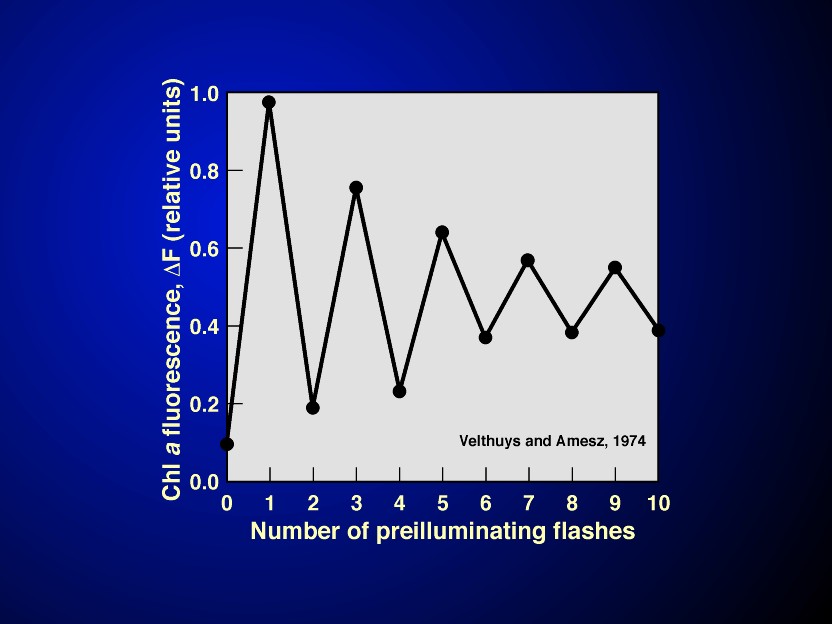
A major advancement took place in the understanding of the functioning of the electron transfer pathway when Velthuys & Amesz (1974) and Bouges-Bocquet (1973) showed that there is a two electron gate on the acceptor side of PSII (see Figure 11 and its legend ); a similar gate was discovered later in anoxygenic photosynthetic bacteria, independently by Vermeglio (1977) and by Wraight (1977). Electrons from the reaction centers are transferred first to a bound quinone molecule QA which is, surprisingly, a one-electron acceptor. The reduced QA transfers its electron to a loosely bound quinone QB; the reduced QB is tightly bound and has a long lifetime; it waits for a second electron until it can be doubly reduced to QB2-. The latter then becomes a quinol (QBH2) after grabbing two protons coming from the side of the membrane it is located on. The electron flow at the QAQB site can be written as (see a review by Crofts & Wraight, 1983):
QAQB+ light-------> QA -QB-----> QAQB- (8)
QAQB- + light ------> QA QB2- (9)
QA QB2-+ 2H+------> QA QBH2 (10)
QA QBH2 + PQ-------> QAQB + PQH2. (11)
Although it is not certain, especially in plants, whether the protonation steps occur before the electron flow or vice versa, the above steps are referred to as the "two electron gate".
The bicarbonate
effect. I have had a personal interest in this
two electron "gate". Over the past twenty-five years or
more, we have been trying to understand the role of bicarbonate
in the reactions on the two photosystems, a phenomenon discovered
by Warburg & Krippahl (1960). Warburg (1964) took this
phenomenon to suggest that oxygen in photosynthesis originated in
CO2, not in water, contradicting the existing idea that oxygen
originated in water. He argued that (1) the 18O experiments of
Ruben et al. (1941) could not be trusted because of equilibration
with both water and CO2; (2) the hypothesis of C.B. van Niel was
only from comparative biochemistry between oxygenic and
anoxygenic organisms, and thus had no definitive value; and (3)
the requirement of bicarbonate in the Hill reaction showed that
it cannot be taken to prove that CO2 was not the source of oxygen
in photosynthesis. I note here parenthetically that the discovery
of the Hill reaction (see Hill & Scarisbrick, 1940), i.e.,
reduction of an artificially added electron acceptor and
evolution of oxygen by chloroplasts, was itself a major finding
that allowed tremendous number of biochemical studies that ensued
from that time; it opened the field to look under the
"hood" of the car, so to say. These were the arguments
that fascinated me. I presented them to a photosynthesis course I
was teaching in the early seventies; it was attended, among
others, by Prasanna Mohanty and Alan Stemler, my PhD students.
Alan jumped at the idea of working on the exciting project of
attempting to understand the role of bicarbonate in the Hill
reaction. Our first paper (Stemler & Govindjee, 1973)
suggested that bicarbonate may function on the water side of
photosynthesis. However, very soon we discovered a major effect
on the acceptor side of photosystem II. We located a role on the
electron acceptor side, specifically at the two-electron gate
(Wydrzynski & Govindjee, 1975; Govindjee et al., 1976;
Jursinic et al., 1976; Khanna et al., 1977, 1981; Blubaugh &
Govindjee, 1988; Eaton-Rye & Govindjee, 1988; Xu et al.,
1990; Govindjee, 1993, and, Govindjee & van Rensen, 1993).
Bicarbonate seems to be involved not so much in the electron flow
per se, but in the protonation steps (also see van Rensen
et al., 1988). Interestingly, this effect is absent in anoxygenic
photosynthetic bacteria (see e.g., Shopes et al.,.1988; Wang et
al., 1992). Differences between photosynthetic bacteria and
photosystem II are, we believe, due to a preponderance of
positively charged amino acids in the QB binding region of PSII
over that in photosynthetic bacteria (Xiong et al., 1996;
1998a). This may be responsible for different protonation
channels in anoxygenic photosynthetic bacteria and in PSII. This
bicarbonate site seems close to (Khanna et al., 1981) , but not
identical to that where certain herbicides bind and act
(Velthuys,1981; Wraight ,1981). Terbutryn inhibits and
kills plants by displacing QB, and bicarbonate seems to be
involved in the protonation of the reduced form of QB. One of the
binding sites of bicarbonate has been shown to be on the non-heme
iron ( see e.g., Diner et al., 1991) and the other near an
arginine residue, D1-R-257 (Xiong et al., 1998b). It is not yet
clear how this acceptor side binding (see Figure
12) would provide another major bicarbonate action on the
oxygen evolving complex expounded by O. Warburg, H. Metzner and
A. Stemler (see Stemler, 1982), and now being
investigated in great detail by Klimov & coworkers
(Allakhverdiev et al., 1997; Hulsebosch et al., 1998; Klimov et
al., 1997; Yruela et al., 1998). It calls for further research.
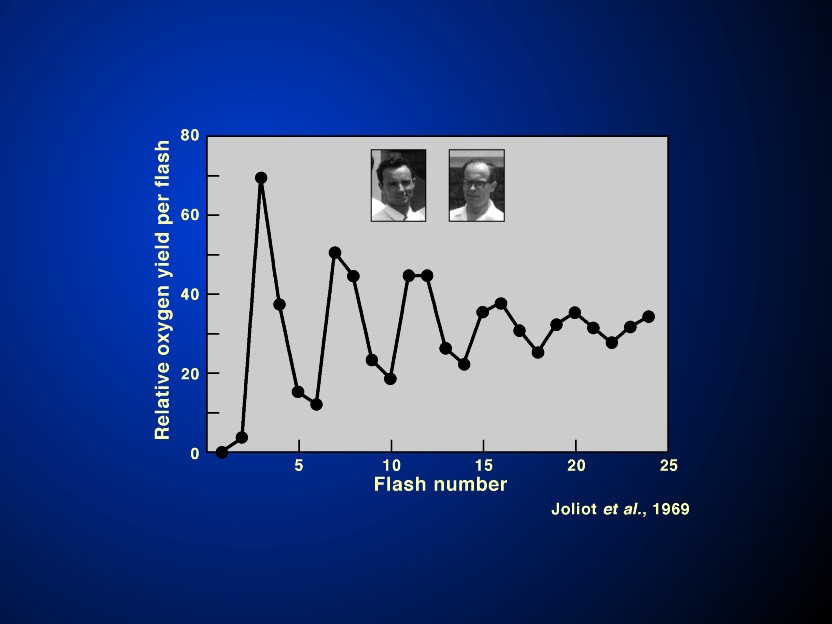
The answer to the question of the minimum number of quanta needed to evolve one oxygen molecule was solved in favour of 8-10 (Emerson & Lewis, 1943; Emerson, 1958; R. Govindjee et al., 1968) rather than of 3 to 4 (Warburg & Negelein, 1923). This is in agreement with the scheme that two light reactions are needed for oxygen evolution and NADP+ reduction. A major breakthrough in the oxygen evolution steps was the discovery by Joliot et al. (1969) that oxygen/flash as a function of flash number oscillates with a period of 4 indicating accumulation of 4 positive charges on some intermediate before water is oxidized to molecular oxygen (Figure 13). A theory evolved by Kok et al.(1970) became the major framework (for alternate models, see Mar & Govindjee, 1972), where the system started mostly in the S1 state in darkness, and oxygen release took place when S4 was converted to S0 (S representing the redox state of the tetranuclear Mn complex):
` S1 + light flash #1; or #5---> S2 (12)
S2 + light flash # 2; or #6---->S3 (13)
S3+ light flash # 3; or # 7----> S4 (14)
S4 + 2H2O-----> S0+ O2 + 4 H+ (15)
S0 + light flash # 4; or # 8---> S1. (16)
The elegant measurements of Dismukes & Siderer (1980) on
multiline EPR (electron paramagtnetic resonance) signals of Mn,
and those of M. Klein and coworkers (see Yachandra et al., 1996;
Roelofs et al., 1996) on EXAFS (Extended X-ray Absorption Fine
Structure) and XAS ( X-ray Absorption Spectroscopy) led to the
identification of the S-state intermediates with mixed valence Mn
complex (a tetranuclear Mn/PSII). The S0 is suggested to be Mn
(II), Mn (III), Mn (IV), Mn (IV) (alternatively, Mn (III), Mn
(III), Mn (III), Mn (IV)); S1 to be Mn (III), Mn (III), Mn (IV),
Mn (IV); and both S2 & S3 as Mn (III), Mn (IV), Mn (IV), Mn
(IV), Mn (IV). The mechanism of water oxidation to
dioxygen is not yet known; however, there are theories as to how
this occurs. I refer the readers to read Renger (1997) and
Babcock & co-workers (Hoganson& Babcock,1997; Tommos
& Babcock, 1998) to obtain a glimpse of the current and two
different thoughts on the mechanism of oxygen evolution. The
recent finding of Messinger et al. (1997a) that S0 is also
paramagnetic will further enhance our ability to understand the
transitions of the S states, presented above. In addition,
the possibility of showing that the S states can be reduced to
S(-3) state lends credence to the current picture of the valence
state of the Mn in the water oxidase (see Messinger et al.,
1997b). On the personal side, I want to mention that Coleman
& Govindjee (1987) were the first to suggest that the Mn
atoms of the Mn cluster were bound to the reaction centre
proteins D1 & D2; and, Kambara et al. (1985) and Padhye et
al. (1986) were the first to suggest that an organic radical,
specifically a histidine, may be the charge accumulator in
addition to Mn during S2 to S3 transition. The role of histidine
is being actively investigated in several laboratories; first, it
seemed evidence was obtained, then, it was challenged; now, I am
waiting for the final judgement before citing any paper on this
topic. Following the early pioneering work of W.F.J. Vermaas and
of B. Diner and coworkers, Bowlby et al. (1996) have reported
that Glutamic-65 and Histidine 337 on D1 protein may be ligands
to Mn; and, Aspartic 103 and Glutamic 104, also on D1, may be
ligands to Calcium. On the other hand, T. Wydrzynski and
coworkers (Messinger et al., 1995) are providing new information,
using elegant mass spectroscopic methods, on the exchangeable
water molecules at the site of water oxidation. Evidence for the
proximity of a Calcium to the Mn cluster, discussed above, has
now been implied from X-ray absorption spectroscopy (Latimer et
al., 1995). Oxygen, the by-product of water oxidation, is
released through the operation of the "oxygen clock"
that involves 4 Mn atoms/PSII (see a detailed earlier review by
Debus, 1992).
A major tool to study the S-states was shown to be
thermoluminescence, a phenomenon discovered by W. Arnold ( see
reviews by Sane and Rutherford, 1986; Vass & Govindjee, 1996).
Thermoluminescence (TL) is light emission, that originates mainly
from PSII. If one shines light on a photosynthetic material, and
freezes to 77K, all the separated charges are frozen, and heating
the sample in darkness leads to thermoluminescence bands
originating from the back reactions (i.e., recombination of
charges) of say, e.g., S2 with QB- or S2 with QA-, etc. P.V.
Sane, in collaboration with V.G. Tatake and T.S. Desai, was
responsible for placing India on the World map of pioneering
research in this area of thermoluminescence. The design of the
instrument at the Bhabha Atomic Research Centre (BARC), Mumbai,
India, where this work was done, provided very high Signal/Noise
ratio. Several (6 or 7) TL bands were fully separated from each
other and these bands, through separation of pigment systems I
and II and through the ingenious use of inhibitors and electron
carriers, were related to specific reactions in PSII and even PSI
(see Sane & Rutherford, 1986; and Vass & Govindjee, 1996,
for reviews). It was because calculations, by Tatake et al.
(1981), of the activation energies of the various TL bands did
not match the Randall-Wilkins theory that I approached Don
DeVault for help; this led to the formulation by DeVault et al.
(1983) of the most appropriate theory for TL in plants. Without
the leadership provided by P.V. Sane, researches on TL at BARC
would have been in directions different from those on the
photosynthetic systems. Relationship of the S-states to the TL
was, however, provided by Inoue &Shibata (1978) when
they discovered the period 4 oscillation in TL and related it to
oxygen evolution steps (see reviews by Vass & Inoue, 1992;
and Inoue, 1996). I refer the readers to the cited reviews to
discover for themselves the discoveries of A.W. Rutherford, G.
Renger, and the research group in Szeged, Hungary without whose
work, TL will not be where it is today. Currently, the TL method
is being used to monitor and characterise site-directed mutants
both on the acceptor (see e.g., Mäenepää et al., 1995) as well
as the donor side of PSII (see e.g., Kramer et al., 1994).
ATP is the energy currency of life. Production of glucose (food) from CO2 requires both NADPH and ATP. Below, we will discuss the breakthroughs that led to the understanding of ATP synthesis: chemiosmotic hypothesis; and the alternate binding site hypothesis.
After indications in several laboratories that algal cells produce ATP in light, Arnon et al. (1954) and Frenkel (1954) discovered photophosphorylation in chloroplasts of plants, and chromatophores of anoxygenic photosynthetic bacteria, respectively. This was a major breakthrough and it took many years to recognize that only in some anoxygenic photosynthetic bacteria, almost all light energy is first converted into ATP energy and then this energy is used for the reversed electron flow to produce the reducing power in the form of NADH. In oxygenic organisms, however, this is not the case as correctly expressed by Rabinowitch (1956). As we know today, ATP synthesis follows electron transport steps that first store energy temporarily by creating a proton motive force; this energy would be otherwise lost if not used for ATP formation (see the scheme of Hill & Bendall, 1960). P. Mitchell (1961), who received the Nobel prize in Chemistry in 1978, provided the theory that a proton motive force ( that is a sum of a pH gradient and a membrane potential) is the energy source of ATP synthesis. Membranes are normally impermeable to protons; and, protons are transferred from one side of the membrane to the other by virtue of the alternate electron and hydrogen atom transfers due to specific location of the electron and hydrogen atom carriers and the energetics of the light-induced electron transfers in photosynthesis. An early breakthrough was the independent experiment of Shen & Shen (1962) and of Hind & Jagendorf (1963) showing that light forms some entity (XE) that can be used later in darkness to produce ATP. Hind & Jagendorf showed that XE is a pH gradient that drives ATP synthesis. Jagendorf & Uribe (1966) showed that ATP can be synthesized from pH gradient created by acid-base transition in total darkness. These experiments, along with those of the others, provided major evidence for the chemiosmotic hypothesis of Mitchell. In photosystem II, water oxidation complex that liberates protons is on the inner side (the lumen) of the thylakoid membranes. Further, plastoquinone is reduced to plastoquinol on the outer side (stroma side) of the thylakoid membrane; the plastoquinol (a hydrogen atom carrier) moves towards Rieske Iron sulfur and cytochrome f (both are electron, not hydrogen atom carriers) that are located towards the lumen side. Here, plastoquinol delivers its electrons to the Rieske Iron sulfur centre and the cytochrome f, leaving the protons to be released into the lumen. This arrangement, thus, allows natural proton translocation from the stroma to the lumen side as the electron transport takes place in PSII. This adds to the pH gradient to be used for ATP synthesis. The equivalence of pH gradient and membrane potential in synthesizing ATP was shown by the elegant experiments of Gräber et al. (1984) and Hangarter & Good (1982) when they varied one keeping the other constant and showing that it was the sum of the two that was important for initiating ATP synthesis.
The understanding of the mechanism by which ATP synthesis
takes place at the ATP synthase using the proton motive force has
been influenced by the following three breakthroughs. (1) The
binding change hypothesis of P. Boyer and co-workers (see Boyer
et al., 1973; and a review by Boyer, 1997; also see Figure 14 and its legend) that suggests that at
one time (a) ADP and Pi (inorganic phosphate) bind weakly at one
site of the alpha-beta pair of the F1 part of the ATP synthase
enzyme; (b) bound ATP is formed from bound ADP and Pi, without
the use of energy at a second alpha-beta pair; and (c) the pH
gradient energy is converted into rotational energy, mainly of
the gamma subunit that extends upto the third alpha-beta pair,
that is used to flip off the ATP free from the third alpha-beta
binding site. These three sites alternate in time. (2) The
rotation feature of the Boyer model has been elegantly
demonstrated directly by fluorescence microscopy and by
photoselection and other experiments (see Capaldi, R.A. 1994;
Duncan et al., 1995. Sabbert et al., 1996, 1997; Junge et al.,
1997) (3) Atomic resolution structure (Abrahams et al., 1994) of
beef-heart mitochondrial F1 showed that the gamma subunit indeed
looks through the alpha-beta pairs; and in agreement with the
Boyer model, the structure shows one alpha-beta pair site empty
(as if ATP was released); another with bound ADP and Pi; and the
third with an equivalent of bound ATP. I consider it highly
likely that the plant, algal, and cyanobacterial ATP synthase
will be basically similar to that of the cow.
CONCLUDING REMARKS
In my opinion, the mechanism of photosynthesis has been probed
by several means. Its face has changed by experiments of many
investigators. I restrict my personal list to deal mainly with
oxygen evolution. It includes, among others, the experiments of
my first mentor Robert Emerson (Emerson & Arnold,1932). U
sing the repetitive flash method and Warburg’s manometry,
they led to the concept of "Photosynthetic Unit" where
excitation energy, absorbed by hundreds of antenna pigment
molecules, is transferred to the reaction center chlorophyll
molecules, the "photoenzyme" or the "energy
trap" for chemistry. Next, was the discovery of the
"Red Drop" (Emerson & Lewis, 1943) in the quantum
yield action spectrum of oxygen evolution in the green alga Chlorella
pyrenoidosa, and the enhancement effect of certain
wavelengths of light on the yield of oxygen evolution in the
"red drop" region (Emerson et al., 1957), discovered by
the use of an excellent monochromator and state-of-the art
manometry; it lead to the concept of two-light reaction and
two-pigment system mechanism of electron transport. It became
well-known due to the working hypothesis of Hill & Bendall
(1960) and became a scientific fact by the experiments of Duysens
et al. (1961) on the antagonistic effect of light absorbed in
pigment system I and II on the redox state of cytochrome f.
The important concept that there is an "oxygen clock",
where four positive charges must accumulate before water can be
oxidized to oxygen was enshrined before us by (a) the experiments
of Joliot et al. (1969) on the periodicity of four in the plots
of the amount of oxygen evolved per flash as a function of flash
number, and (b) the so-called "S-states" model of
charge accumulation enunciated by Kok et al. (1970). Such a
periodicity of four, reflecting indirectly the S-states, has also
been observed in chlorophyll a fluorescence (Delosme,
1971; Joliot & Joliot, 1971), and recently, Shinkarev et al.
(1997) have even managed to obtain the kinetics of the last step
of oxygen evolution from analysis of the data on the decay of
chlorophyll a fluorescence in single flashes of light.
Thermoluminescence, discovered by W. Arnold & H. Sherwood
(see Arnold, 1991), that has been exploited both by P.V. Sane
(Sane & Rutherford, 1986) as well as myself (DeVault et al.,
1983; Vass & Govindjee, 1996), has uniquely probed the
characteresteristics of the S-states of oxygen evolution (Inoue
& Shibata, 1978; Inoue, 1996). The most elegant probes for
showing that Kok’s S-states are Manganese are the low
temperature EPR (Dismukes& Siderer, 1980); and the Extended
X-ray Absorption Fine Structure (EXAFS) spectroscopy (M. Klein,
K. Sauer & co-workers; see Yachandra et al., 1996). At the
end, I do want to emphasise two elegant conceptual work on
another area, that of phosphorylation: (a) the chemiosmotic
theory of P. Mitchell in which proton motive force across a
membrane provide energy for ATP synthesis (Mitchell, 1961) and
(b) the elegant theory of Paul Boyer as to how this comes about:
by conversion of electrochemical energy to conformational energy
(see Boyer, 1977). Finally, I wish to mention that mimicking
photosynthesis in vitro has been a dream of many, and the
recent success of Steinberg-Yfrach et al. (1998) in making
"lots of ATP" in artificial liposome membranes,
energised by a synthetic system (carotene-porphyrin-quinone, is
highly commendable. It would have made my second mentor Eugene
Rabinowitch quite happy.
Abrahams, J.P., Leslie, A.G.W., Lutter, R.& Walker, J.E. 1994. Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature 370: 621-628.
Allakhverdiev, S., Yruela, I., Picorel, R.& Klimov, V.V.1997.Bicarbonate is an essential constituent of the water oxidizing complex of photosystem II. Proc. Nat. Acad. Sci. U.S.A. 94: 5050-5054.
Amesz,J. 1996. The heliobacteria, a new group of photosynthetic bacteria. J. Photochem. Photobiol. B-Biol. 30: 89-96.
Arnold, W. (1991). The experiments. Photosynth. Res. 27: 73-82.
Arnon D.I. 1955. The chloroplast as a complete photosynthetic unit. Science 122: 9-16. Arnon, D.I., Allen, M.B & Whatley, F.R.1954. Photosynthesis by isolated chloroplasts. Nature 174: 394-396.
Bassham, J.A., Benson, A.A., Kay, L.D., Harris, A.Z., Wilson, A.T. & Calvin, M. 1954. The path of carbon in photosynthesis. XXI. The cyclic regeneration of carbon dioxide acceptor. J. Amer. Chem. Soc. 76: 1760-1770.
Blubaugh, D. & Govindjee. 1988. The molecular mechanism of the bicarbonate effect at the plastoquinone reductase site of photosynthesis. Photosynth. Res. 19: 85-128.
Bouges-Bocquet, B. 1973. Electron transfer between the two photosystems in spinach chloroplasts. Biochim. Biophys. Acta 314: 250-256.
Bowlby, N.R., Sithole, I., Babcock, G.T. & McIntosh, L. 1996. Potential metal ligands in photosynthetic water oxidation identified by site directed mutagenesis of Synechocystis sp pCC 6803. Ber. Bunsens Geselschaft Phys Chem-Chem. Phys. 100: 1978-1986.
Boyer, P.D. 1997. The ATP synthase- a splendid molecular machine. Ann. Rev. Biochem. 66: 717-749.
Boyer, P.D., Cross, R.L. & Momsen, W. 1973. A new concept for energy coupling in oxidative phosphorylation based on a molecular explanation of the oxygen exchange reactions. Proc. Nat. Acad. Sci. U.S.A. 70: 2837-2839.
Brockmann, H. & Lipinski, A. 1983. Bacteriochlorophyll g. A new bacteriochlorophyll from Heliobacterium chlorum. Arch. Microbiol. 136: 17-19.
Brody, S. S.& Rabinowitch, E. 1957. Excitation lifetime of photosynthetic pigments in vitro and in vivo. Science 125: 555..
Butler,W. L. 1962. Effects of red and fared light on the fluorescence yield of chlorophyll a in vivo. Biochim. Biophys. Acta 64: 309-317.
Calvin, M. 1989. Forty years of photosynthesis and related activities. Photosynth. Res. 21: 3-16.
Capaldi, R.A. 1994. F1-ATPase in a spin. Struct. Biol. 1: 660-663.
Cleland, W.W., Andrews, T.J., Gutteridge, S., Hartman, F.C. & Lorimer, G. 1998. Mechanism of Rubisco: The carbamate as general base. Chem. Reviews 98: 549- 562.
Coleman, W. & Govindjee. 1987. A model for the mechanism of chloride activation of oxygen evolution in photosystem II. Photosynth. Res. 13: 199-223.
Coleman, W.J., Mattioli, T.A., Youvan, D.C. & Rutherford, A.W. 1997. Site directed mutations near the L-subunit D-helix of the purple bacterial reaction center: A partial model for the primary donor of photosystem II. Biochemistry36 : 2178-21??
Collyer, C.A., Guss, J.M., Sugimura, Y., Yoshizaki, F.& Freeman, H.C. 1990. Crystal structure of plastocyanin from a green alga, Enteromorpha prolifera.J. Mol. Biol. 211: 617-632.
Crofts, A.R. & Wraight, C.A. 1983. The electrochemical domain of photosynthesis. Biochim. Biophys. Acta 726: 149-185.
Debus, R. 1992. The manganese and the calcium ions of photosynthetic oxygen evolution. Photosynth. Res. 1102: 269-352.
Delosme, R.1971. New results about chlorophyll fluorescence in vivo.In: Forti, G. Avron, M. and Melandri, A. (eds.) Proceedings of the Iind International Congress on Photosynthesis Research, Vol. I, pp. 187-195. Dr. W. Junk Publishers, The Hague.
Deisenhofer, J., Epp, O., Miki, M., Huber, R.& Michel, H. 1984. X-ray structure analysis of a membrane protein complex. Electron density map at 3 A resolution and a model of the chromophores of the photosynthetic reaction centre from Rhodopsuedomonas viridis. J. Mol. Biol. 180: 385-398.
Deisenhofer,J., Epp,O., Miki, K., Huber, R. & Michel, H. 1985. Structure of the protein subunits in the photosynthetic reaction centre of Rhodopsuedomonas viridis at 3 Å resolution. Nature 318: 618-624.
Deisenhofer, J., Epp, O., Sinning, I. & Michel, H. 1995. Crystallographic refinement at 2.3 Å resolution and refined model of the photosynthetic reaction centre from Rhodopsuedomonas viridis. J. Mol. Biol. 246: 429-457.
DeVault, D. 1989. Tunneling enters biology. Photosynth. Res. 22: 5-10.
DeVault, D.& Chance, B. 1966. Studies of photosynthesis using a pulsed laser: Temperature dependency of cytochrome oxidation rate in Chromatium vinosum. Evidence for tunneling. Biophys. J. 6: 825-847.
DeVault, D., Govindjee & Arnold, W. 1983. Energetics of photosynthetic glow peaks. Proc. Nat. Acad. Sci. U.S.A. 80: 983-987.
Diner, B., Petrouleas, V. & Wendoloski, J.J. 1991. The iron-quinone acceptor of photosystem II. Physiol. Plant. 81: 423-436.
Dismukes, C. & Siderer, Y. 1980. Intermediates of a polynuclear manganese center involved in photosynthetic oxidation of water. Proc. Nat. Acad. Sci. U.S.A. 78: 274-278.
Döring, G., Renger, G., Vater, J. & Witt, H.T. 1969. Properties of the photoactive chlorophyll aII in photosynthesis. Z. Naturforsch. 24b: 1139-1143.
Duncan, T.M., Bulygin, W., Zhou, Y., Hutcheson, M.L. & Cross, R.L. 1995. Rotation of subunits during catalysis by Escherichia coli F1-ATPase. Proc. Nat. Acad. Sci. U.S.A. 92: 10964-10968.
Duysens, L.N.M. 1952. Transfer of excitation energy in photosynthesis. Ph.D. dissertation. State University at Utrecht, The Netherlands.( Drukkerij en Uitgevers- maatschappij v/h Kemink en zoon N.V. , Domplein 2, Utrecht)
Duysens, L.N.M. 1989. The discovery of the two photosynthetic systems: A personal account. Photosynth. Res. 21: 61-79.
Duysens, L.N.M. & Sweers, H.E. 1963. Mechanism of the two photochemical reactions in algae as studied by means of fluorescence. In Japanese Society of Plant Physiologists (eds) Studies on Microalgae and Photosynthetic Bacteria:: 353-372. University of Tokyo Press, Tokyo.
Duysens, L.N.M., Amesz, J. & Kamp, B.M. 1961. Two photochemical systems in photosynthesis. Nature 190: 510-511.
Eaton-Rye, J.J. & Govindjee. 1988. Electron transfer through the quinone acceptor complex of photosystem II after one or two actinic flashes in bicarbonate-depleted spinach thylakoid membranes. Biochim. Biophys. Acta 935: 248-257; also see pp. 237-247.
Emerson, R. 1958. The quantum yield of photosynthesis. Ann. Rev. Plant Physiol. 9:1- 24.
Emerson, R. & Arnold, W.A. 1932a. A separation of the reactions in photosynthesis by means of intermittent light. J. Gen. Physiol. 15: 391-420.
Emerson, R. & Arnold, W.A. 1932b. The photochemical reaction in photosynthesis J. Gen. Physiol. 16: 191-205.
Emerson, R. & Lewis, C.M.1943. The dependence of quantum yield of Chlorella photosynthesis on wavelength of light. Am J Bot 30: 165-178
Emerson, R. & Rabinowitch, E. 1960. Red drop and role of auxiliary pigments in photosynthesis. Plant Physiol. 35: 477-485.
Emerson, R., Chalmers, R.V. & Cederstand, C. 1957. Some factors influencing the long-wave limit of photosynthesis. Proc. Nat. Acad. Sci. U.S.A. 43: 133-143.
Feher, G. 1998. Three decades of research in bacterial photosynthesis and the road leading to it- a personal account. Photosynth. Res. 55: 3-40.
Feher, G., Allen, J.P., Okamura, M. & Rees, D.C. 1989. Structure and function of bacterial photosynthetic reaction centres. Nature 339: 111-116.
Fiener, R. & Huber, R. 1993. Refined crystal structure of phycoerythrin from Porphyridium cruentum at 2.3 Å resolution and localization of the g subunit. Eur. J. Biochem. 218: 103-106.
Fenna, R.E. & Mathews, B.W. 1976. Structure of a bacteriochlorophyll a protein from Prosthecochloris aestuarii. Brookhaven Symp. Biol. 28: 170-182.
Frenkel, A.W. 1954. Light-induced phosphorylation by cell-free preparations of photosynthetic bacteria. J. Amer. Chem. Soc. 76: 5568-5569.
Gaffron, H. & Wohl, K. 1936. Zur Theorie der Assimilation. Naturwissenschaften 24: 81-90; 103-107.
Gest, H. & Favinger, J.L. 1983. Heliobacterium chlorum, an oxygenic brownish-green photosynthetic bacterium containing a 'new' form of bacteriochlorophyll. Arch. Microbiol. 136: 11-16.
Govindjee.1993. Bicarbonate reversible inhibition of plastoquinone reductase in photosystem II. Z. Naturforschung C. 48: 251-258.
Govindjee. 1995. Sixty-three years since Kautsky: Chlorophyll a fluorescence. Aust. J. Plant Physiol. 22: 131-160.
Govindjee & Rabinowitch, E. 1960. Two forms of chlorophyll a in vivo with distinct photochemical function. Science 132: 355-356.
Govindjee & van Rensen, J.J.S. 1993. Photosystem II reaction center and bicarbonate. In Deisenhofer, J. & Norris, J.(eds) The Photosynthetic Reaction Center: 357-389. Academic Press, San Diego.
Govindjee, Ichimura, S., Cederstrand, C. & Rabinowitch, E. 1960. Effect of combining far-red light with shorter wave light on the excitation of fluorescence in Chlorella. Arch. Biochem. Biophys. 89: 322-323.
Govindjee, Pulles, M.P.J., Govindjee, R., van Gorkom, H.J. & Duysens, L.N.M. 1976. Inhibition of the reoxidation of the secondary electron acceptor of photosystem II by bicarbonate depletion. Biochim. Biophys. Acta 449: 602-605.
Govindjee, Amesz, J. & Fork, D.C. 1986. Light Emission by Plants and Bacteria. Academic Press, Orlando.
Govindjee, Knox, R.S. & Amesz, J.(eds).1996. Photosynthetic Unit: Antenna and Reaction Centers. Photosynth. Res. 48: 1-319.
Govindjee, R., Thomas, J.B. & Rabonowitch, E.1961. Second Emerson Effect in the Hill reaction of Chlorella cells with quinone as oxidant. Science 132: 421.
Govindjee, R., Govindjee & Hoch, G. 1962. The Emerson enhancement effect in TPN- photoreduction by spinach chloroplasts. Biochem. Biophys. Res. Comm. 9:222- 225.
Govindjee, R., Govindjee & Hoch, G. 1964. Emerson enhancement effect in chloroplast reactions. Plant Physiol. 39: 10-14.
Govindjee, R., Rabinowitch, E.& Govindjee. 1968. Maximum quantum yield and action spectrum of photosynthesis and fluorescence in Chlorella. Biochim. Biophys. Acta 162: 539-544.
Govindjee, Amesz. & Knox, R.S. (Editors)1996. Photosynthetic unit: antenna and reaction centers. Photosynth. Res. 48: 1-319.
Gräber, P., Junesch, U. & Schatz, G.H. 1984. Kinetics of proton transport coupled ATP synthesis in chloroplasts. Activation of the ATPase by an artificially generated pH and psi. Ber. Bunsenges Physik. Chem. 88: 599-608.
Greenfield, S. & Wasielewski, M.R. 1996. Excitation energy transfer and charge separation in the isolated Photosystem II reaction center. Photosynth. Res. 48: 83- 97.
Greenfield, S., Seibert, M., Govindjee & Wasielewski, M.R. 1997. Direct measurement of the effective rate of primary charge separation in isolated photosystem II reaction center. J. Phys. Chem. 101: 2251-2255.
Groot, M.-L., van Mourik, F., Eickelhoff, C., van Stokkum, I.H.M., Dekker, J.P. & van Grondelle, R. 1997. Charge separation in the reaction center of photosystem II studied as a function of temperature. Proc. Nat. Acad. Sci. U.S.A. 94: 4389-4393.
Hangarter, R. & Good, N. 1982. Energy thresholds for ATP synthesis in chloroplasts. Biochim. Biophys. Acta 681: 397-404.
Hatch, M.D. (Hal).1992. I can’t believe my luck. Photosynth. Res. 33: 1-14.
Hill, R. & Bendall, F. 1960. Function of two cytochrome components in chloroplats: a working hypothesis. Nature 186: 136-137.
Hatch, M.D.. & Slack, C.R..1966. Photosynthesis in sugarcane leaves: A new carboxylation reaction and the path of sugar formation. Biochem. J. 101: 103-111.
Hill, R. & Scarisbrick, R.1940. Production of oxygen in illuminated leaves. Nature 146: 61-62
Hind, G. & Jagendorf, A.T. 1963. Separation of light and dark stages in photophosphorylation. Proc. Nat. Acad. Sci. U.S.A. 49: 715-722.
Hoff, A. & Amesz, J. (eds). 1995. Energy transfer and stabilization in plants and bacterial photosynthetic reaction centers. Chemical Physics194 (Nos. 2 & 3).
Hoff, A. & Deisenhofer, J. 1997. Photophysics of photosynthesis- structure and spectroscopy of reaction centers of purple bacteria. Physics Reports 287: 2-247.
Hofmann, E., Wrench, P.M., Sharples, F.P., Hiller, R.G., Welte, W. & Dietrichs, K.1996..Structural basis of light harvesting by carotenoids- peridinin- chlorophyll protein from Amphidinium carterae.Science 272: 1788-1791.
Hoganson, C.W. & Babcock, G.T. 1997. A mettaloradical mechanism for the generation of oxygen from water in photosynthesis. Science 277: 1953-1956.
Hulsebosch, R.J., Allakhverdiev, S.I., Klimov, V.V., Picorel, R. &..Hoff, A.J.. 1998. Effect of bicarbonate on the S-2 multiline EPR signal of the oxygen evolving complex in photosystem II membrane fragments. FEBS Lett 424: 146-148.
Huzisige, H. & Ke, B.1993. Dynamics of the history of photosynthesis. Photosynth. Res. 38: 185-209.
Inoue, Y. 1996. Photosynthetic thermoluminescence as a simple probe of photosystem II electron transport. In: J. Amesz & A.J. Hoff, Eds.Biophysical Techniques in Photosynthesis. Pp. 93-108. Kluwer Academic, Dordrecht, The Netherlands.
Inoue, Y. & Shibata, K. 1978.Oscillation of thermoluminescence at medium low temperature. FEBS Lett 85: 193-197.
Jagendorf, A.T. & Uribe, E. 1966. ATP formation caused by acid-base transition of spinach chloroplasts. Proc. Nat. Acad. Sci. U.S.A. 55: 170-177.
Joliot, P.. & Joliot, A..1971.Studies on the quenching properties of Photosystem II electron acceptor. In: Forti, G. Avron, M. and Melandri, A. (eds.) Proceedings of the Iind International Congress on Photosynthesis Research, Vol. I, pp. 26-38. Dr. W. Junk Publishers, The Hague.
Joliot, P., Barbieri, G. & Chabaud, R. 1969. Un nouveau modèle des centres photochimiques du système II. Photochem. Photobiol. 10: 309-329.
Junge, W., Lill, H. & Engelbrecht, S. 1997. ATP synthase: an electrochemical transducer with rotatory mechanics. Trends Biochem. Sci. 22: 420-423.
Jursinic, P., Warden, J. & Govindjee. 1976. A major site of bicarbonate effect in system II reaction: Evidence from ESR signal IIvf, fast fluorescence yield changes and delayed light emission. Biochim. Biophys. Acta 440: 323-330.
Kambara, T. & Govindjee.1985. Molecular mechanism of water oxidation in photosynthesis based on the functioning of manganese in two different environments. Proc. Nat. Acad. Sci. U.S.A. 82: 6119-6123.
Karpilov, Y.S.1960. Distribution of radioactive carbon amongst products of phortosynthesis of maize. Tansactions Kazan Agricultural Institute 421: 15-24.
Kendall, H.W. (Chair), Beachy, R., Eisner, T., Gould, F., Herdt, R., Raven, P.H., Schell, J.S. & Swaminathan, M.S. 1997. Bioengineering of Crops. Report prepared for Mr. Ismail Serageldin, Vice President of the World Bank and Chairman, Consultative Group on International Agricultural Research, World Bank ` Panel on Transgenic Crops . Environmentally and Socially Sustainable Development Studies and Monograph Series 23, 30 pp; Washington, DC.
Khanna, R., Govindjee & Wydrzynski, T.1977. Site of bicarbonate effect in Hill reaction: Evidence from the use of artificial electron acceptors and donors. Biochim. Biophys. Acta 462: 208-214.
Khanna, R., Pfister, K., Keresztes, A., van Rensen, J.J.S. & Govindjee. 1981. Evidence for a close spatial location of the binding sites of CO2 and for the photosystem II inhibitors. Biochim. Biophys. Acta 634: 105-116.
Klimov, V.V., Hulsebosche, R.J., Allakhverdiev, S., Wincencjusz, H., van Gorkom, H.J. & Hoff, A.J.1997.Bicarbonate may be required for ligation of manganese in the oxygen evolving complex of photosystem II. Biochemistry 36: 16277-16281.
Kok, B. 1956. On the reversible absorption change at 705 mm in photosynthetic organisms. Biochim. Biophys. Acta 22: 399-401.
Kok, B. 1957. Light induced absorption changes in photosynthetic organisms. Acta Botan. Neerl. 6: 316-336.
Kok, B. 1959. Light induced absorption changes in photosynthetic organisms. II. A split beam difference spectrophotometer. Plant Physiol. 34: 184- 192.
Kok, B., Forbush, B., and McGloin, M.1970. Cooperation of charges in photosynthetic oxygen evolution. I. A linear four step mechanism. Photochem. Photobiol. 11: 457-475.
Kortschak, H., Hartt, C.E. & Burr, G.E.1965. CO2 fixation in sugarcan leaves. Plant Physiol. 40: 209-213.
Kramer, D., Roffey, R.A., Govindjee & Sayre, R.T. 1994. The At thermoluminescence band from Chlamydomonas reinhardtii and the effects of mutagenesis of histidine residues on the donor side of the photosystem II D1 polypeptide.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}